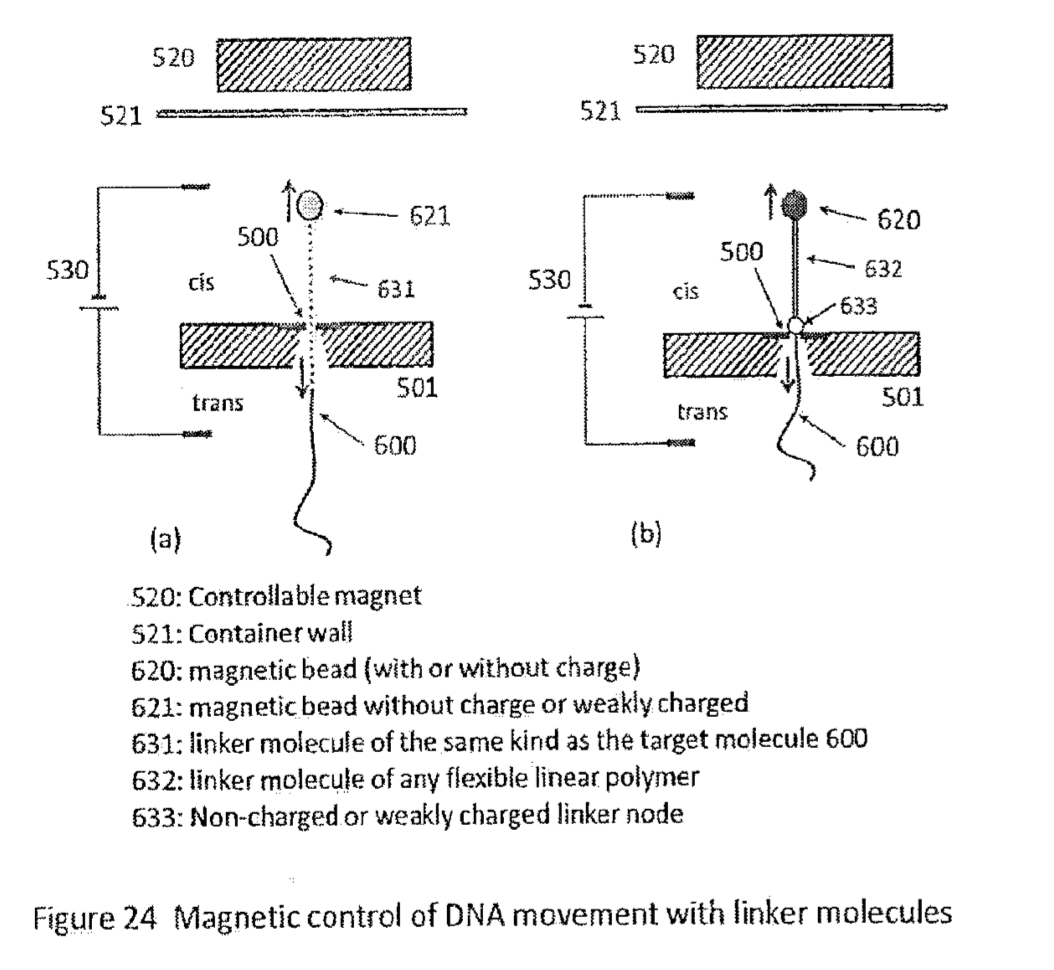
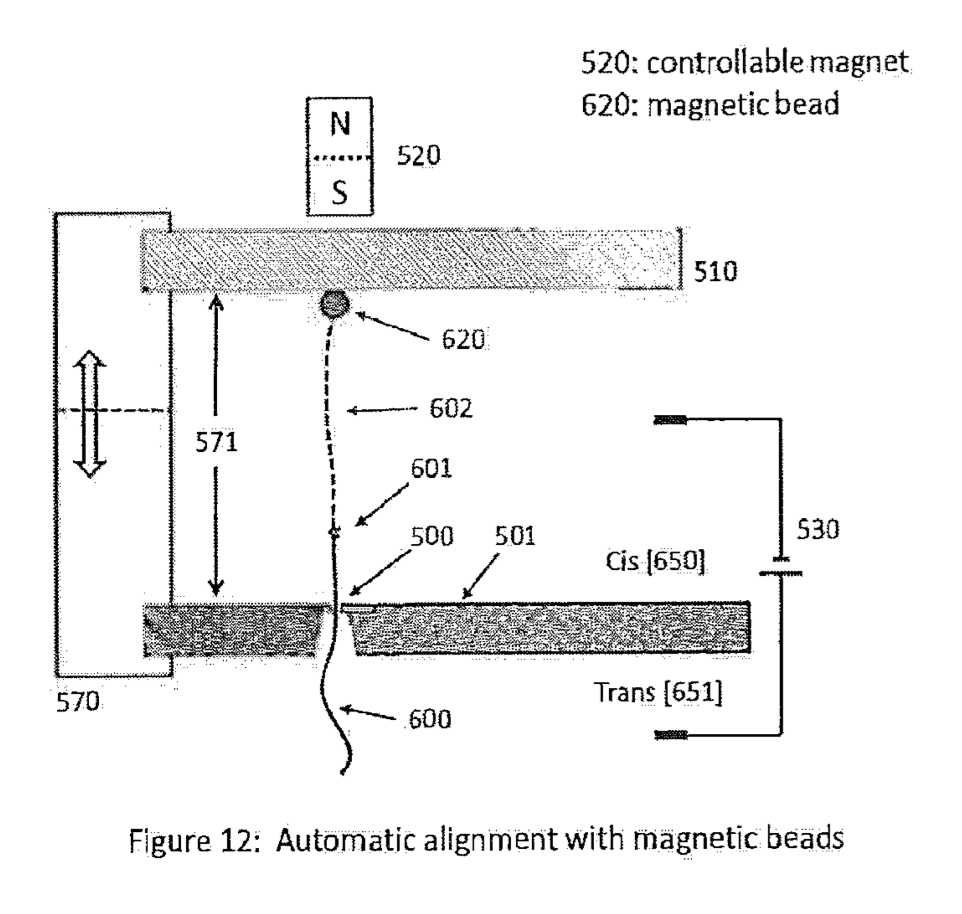
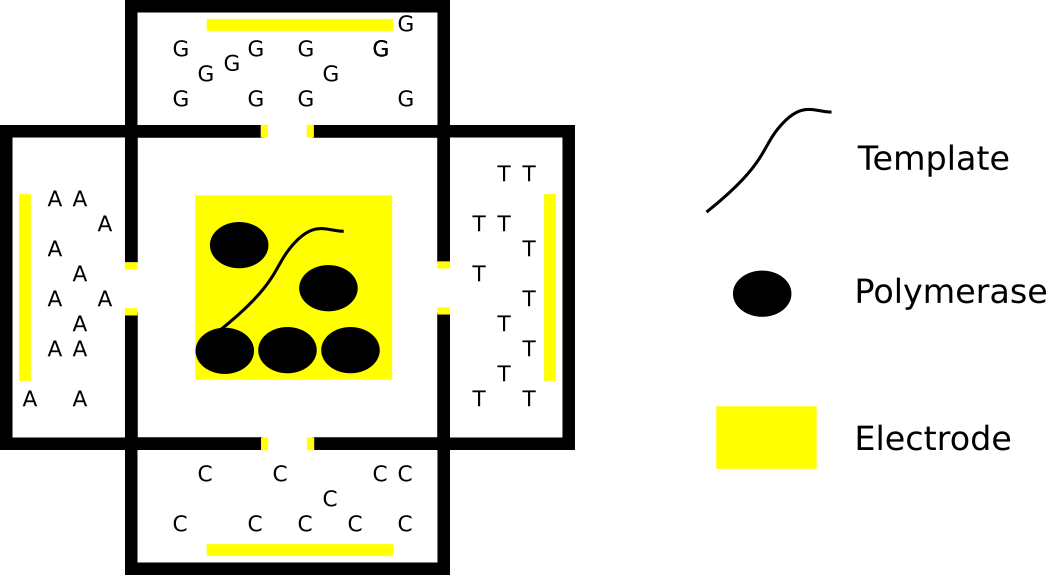
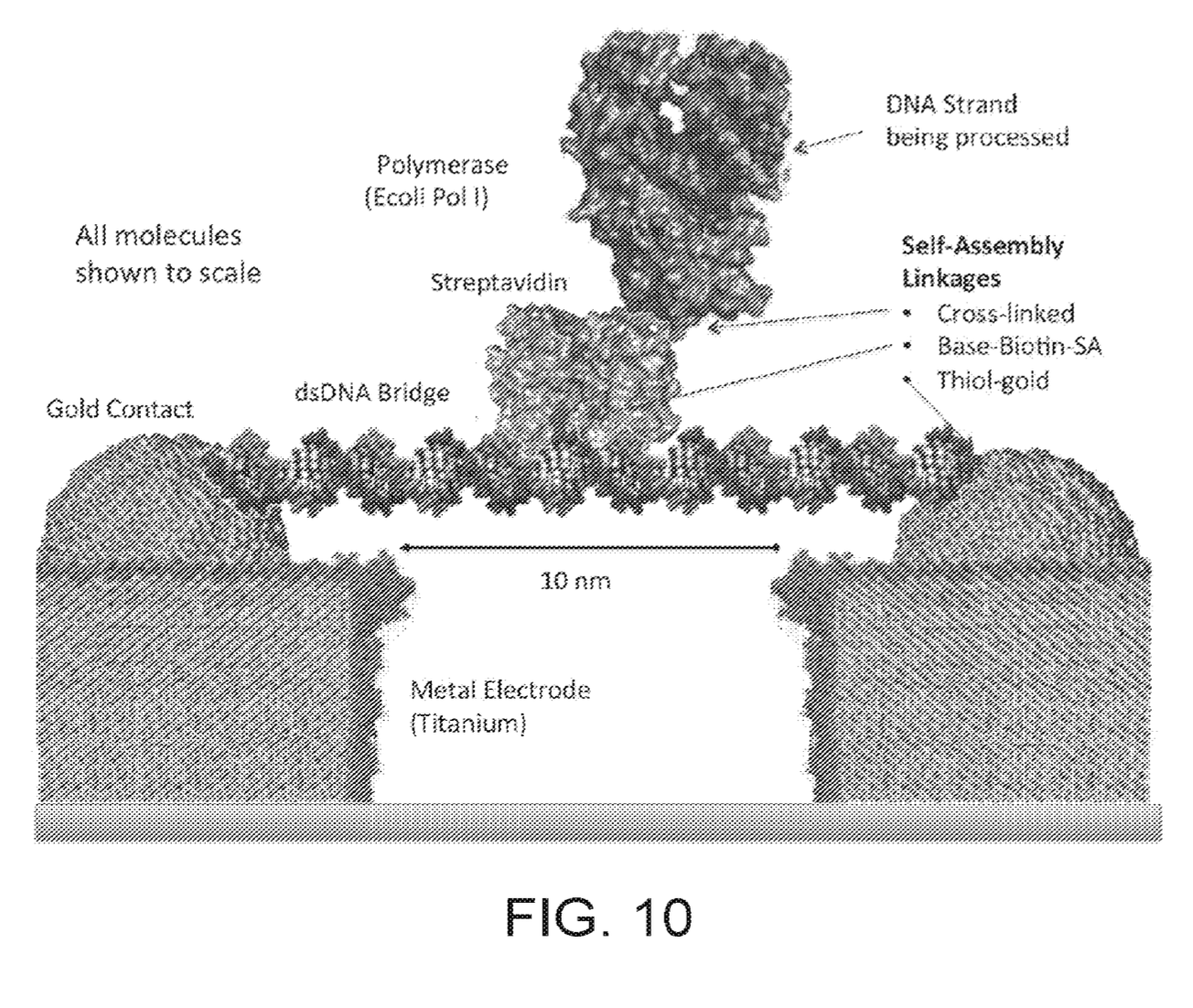
Apton Biosystems
Another day, another sequencing company. This time Apton Biosystems. Checkout the complete list of sequencing companies, for links to all other posts. Unfortunately there’s not much to say about Apton, but this post contains what information seems to be available.
Business
According to their site Apton Biosystems was founded in 2012. SEC filings [1] indicate they raised ~10.5MUSD in 2015. The website lists investors as including Khosla Ventures, Cenova Capital, Samsung Catalyst Fund, and Cowin Venture [5].
From archived copies of their website at the Internet Archive, the site was updated sometime in 2018 (before April?) to provide more information and explicitly mention sequencing (as seemly the focus). Existing patents however only mention sequencing incidentally (by my reading) and focus on protein detection.
Technology
There’s very little information on the Apton sequencing approach, which I would assume is still in development. Current patents do not refer to a specific sequencing approach (for example, sequencing by synthesis or sequencing by hybridisation), and only mention sequencing as one possible application.
The website is more explicit, saying “Apton Biosystems is developing a high throughput system that can sequence the human genome for $10.” [5]. A number of DNA sequencing focused jobs have also been posted [3] [4].
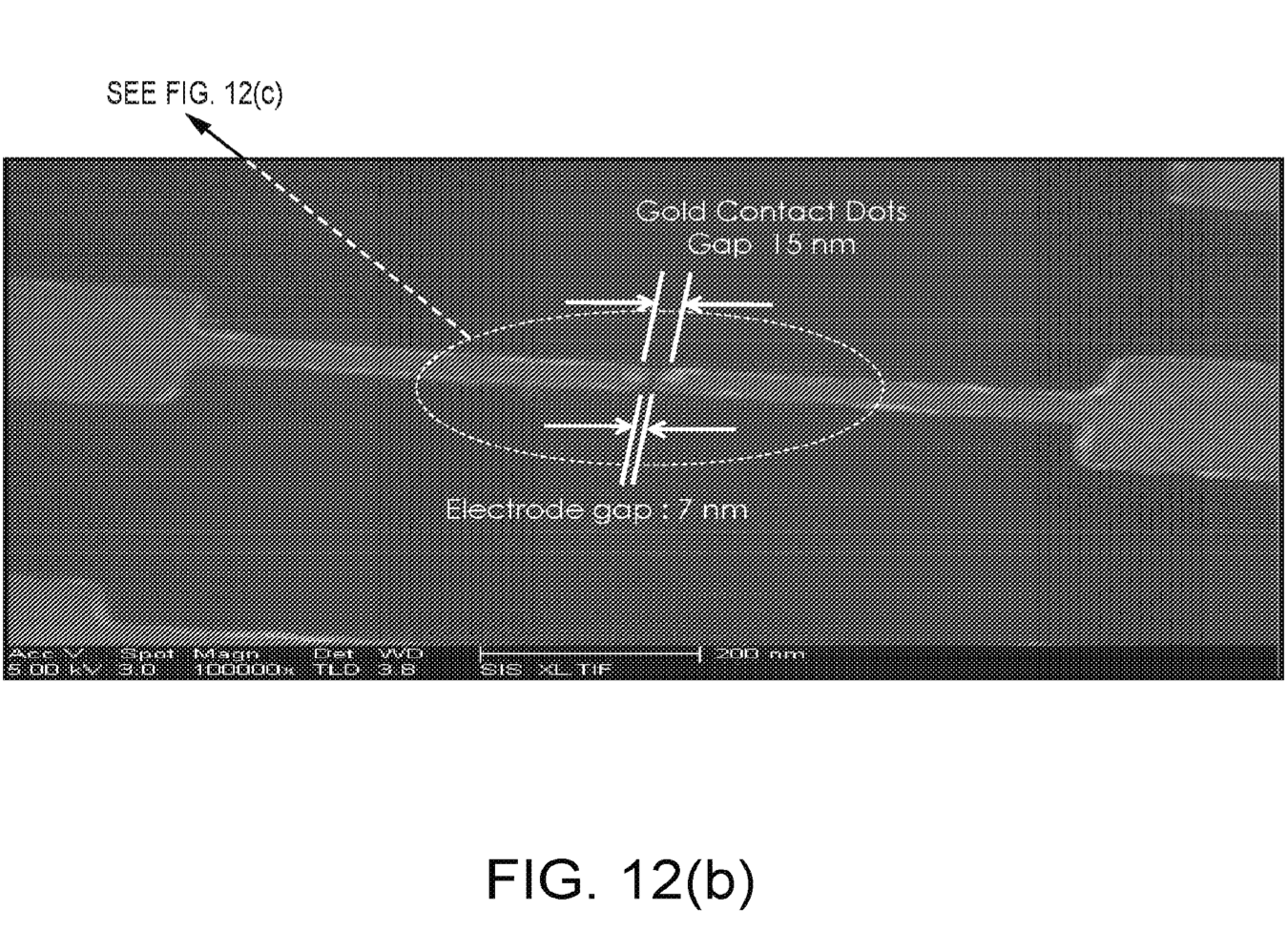
A recent poster provides a little more information [2]. In particular it’s clear that they are developing a single molecule optical approach (like Helicos, Direct Genomes, SeqLL), but 4 color (one would assume one dye per base, but see below). They say the approach can give 20nm resolution, and is therefore a super-resolution approach (beating the diffraction limit). However, there’s still no mention of a particular sequencing chemistry, just that that SNPs were detected using hybridisation probes and single base extension reactions.

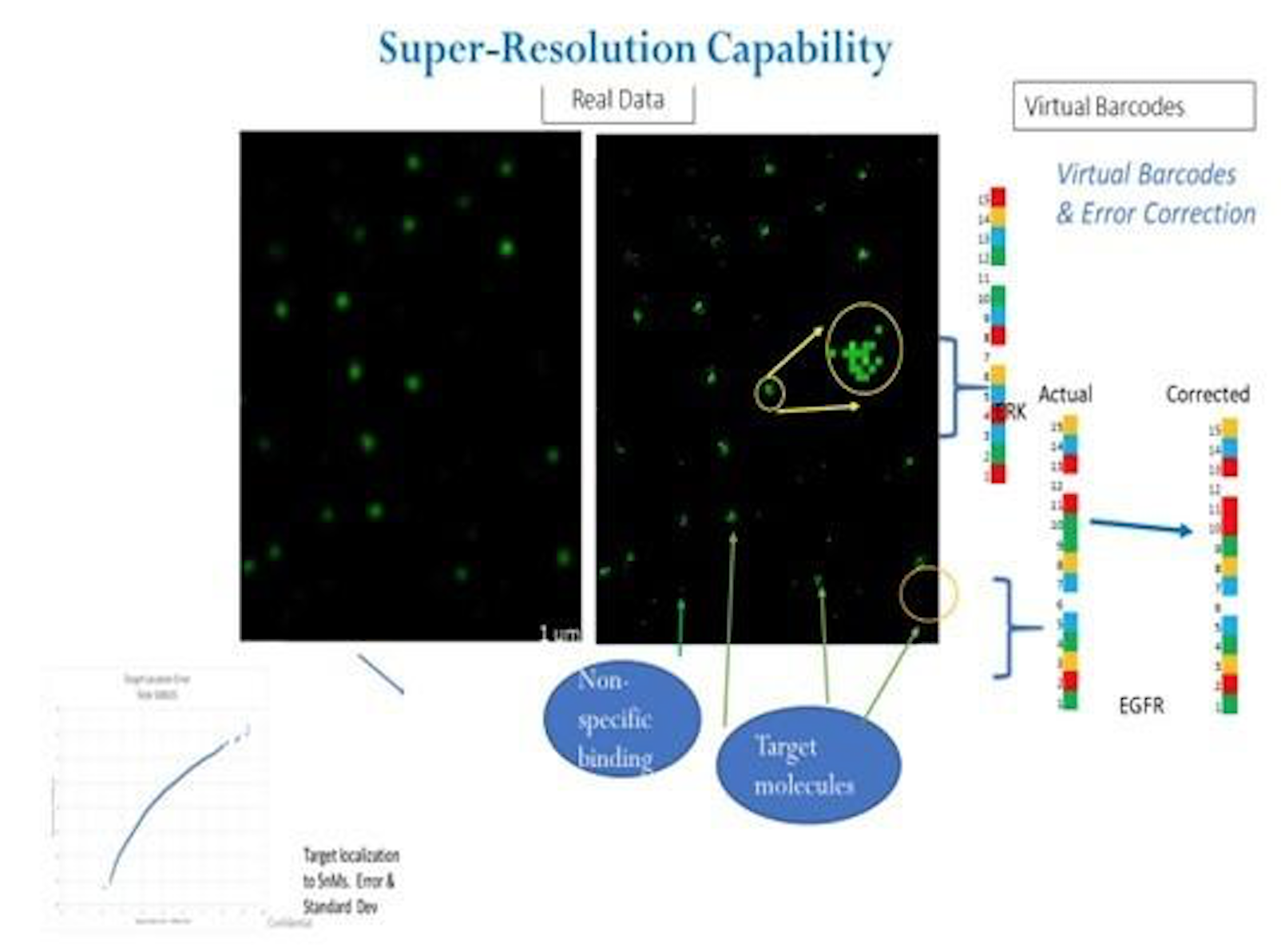
It’s also not clear what super-resolution approach is being used…
In Illumina sequencing cluster positions have long been identified with sub-pixel resolution (you do a gaussian fit around the maximum intensity to get a sub-pixel resolution cluster location) as such it could be said that the Illumina approach can/does have super-resolution qualities.
However, without more details of the sequencing approach used by Apton it’s unclear what advantages this brings.
The poster and patents, also mention error correction, but it’s unclear how they would apply this to sequencing. The use of error correction in the platform, obviously bring to mind Cygnus’ approach.
I’ll be on the look out for more patents, and more information as it appears.
Notes
[1] SEC Filing appear to indicate they raised in the order of 10.5MUSD in 2015: https://www.sec.gov/cgi-bin/browse-edgar?action=getcompany&CIK=0001557398
[2] Poster/talk abstract?
http://cancerres.aacrjournals.org/content/78/13_Supplement/415
“The purpose of this study was to evaluate the system capabilities of a new single-molecule detection platform capable of both genomic and proteomic analysis of cellular pathways using very small amounts of tumor material. The system has 4-color optics, single-fluorophore detection capability, localization of molecules to within a 20nm area, a flow cell with an area of 940 mm2 and the ability to detect > 109 molecules on the surface.”
[3] Job posting
https://www.ventureloop.com/ventureloop/jobdetail.php?jobid=893846&utm_source=joraus&utm_campaign=joraus&utm_medium=organic
“Apton Biosystems Inc. is based in Pleasanton, CA. The company was started in 2012 with the goal of revolutionizing the way cellular processes and pathways are characterized. We are developing an ultra-low-cost and high-throughput sequencing platform based on our 4-color single molecule detection system. This system was developed for the detection and quantification of multiple analytes (DNA, RNA and Proteins) on the same platform and uses proprietary optical and authentication technologies to push the limits of molecular detection and quantification.”
[4] https://www.glassdoor.com/job-listing/lead-data-analyst-bioinformatic-scientist-apton-biosystems-JV_IC1147390_KO0,41_KE42,58.htm?jl=2740301303
“We are developing an ultra-low-cost and high-throughput sequencing platform based on our 4-color single molecule detection system.”
[5] Website Aptionbio.com.
Old/Demo website? ganaraajassociates.
[6]
https://patents.google.com/?assignee=APTON+BIOSYSTEMS+Inc
https://patents.google.com/patent/US20150330974A1
“In one embodiment, the method is computer implemented. In another embodiment, K is one bit of information per cycle. In other embodiments, K is two bits of information per cycle. K can also be three or more bits of information per cycle.”
Long section describing the use of aptamer probes with a long tail. The aptamers are detected by synthesising a complementary strand, and detecting the associated incorporations using an ISFET sensor (this seems odd in an otherwise optical system).