Are there mutations in SARS-CoV-2 CDC qPCR Primer Sites?
I was curious to know if there were any documented mutations which cover CDC Primers/Probes [1]. There’s work that has shown that mismatches in qPCR assays can “completely abolish PCR amplification” [2]. For diagnostic applications, mutations could mean that a qPCR based test would fail to detect SARS-CoV-2 or result in reduced sensitivity.
So, I downloaded all replacements (amino acid substitutions) from CoV-GLUE [3] [4]. I then extracted the nucleotide location identified in each replacement [5]. I then removed any duplicate locations. this resulted in a total of 3527 locations.
I then extracted the CDC primer sequences [6]. I wrote a small tool to do the following:
- Load in the reference sequence, create a new sequence indicate mutation locations on the reference.
- Find the location of the primer sequences on the reference [7].
- For each primer, note where on the primer sequence there are mutations in the reference.
- Report mutation location on primer.
Mismatches near the 3′ end appear to be more significant, I’ve therefore plotted the number of mutations, based on there distance from the 3′ end of the primer. The plot below looks at primers only:
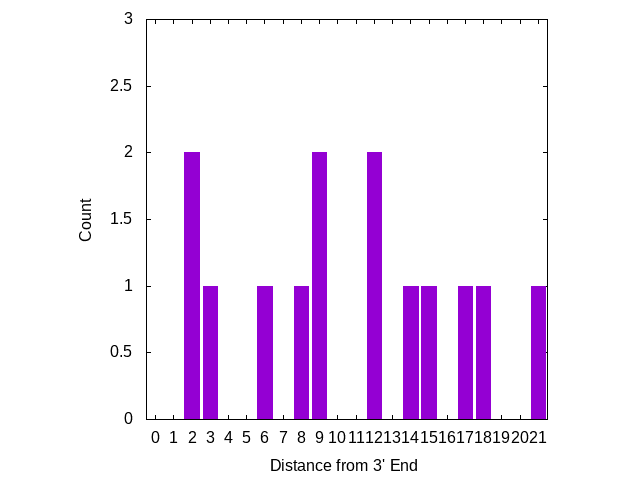
Three mutations are in locations which “may result in a 658-fold underestimation of initial copy number” [2] [8]. But there are no mutations on the 3′ terminal base, where a mismatch is likely to “abolish amplification”.
There does appear to be one mutation in the 3′ terminal base of one of the probes. However, I suspect terminal probe mutations are less significant than those in primers.
This analysis excludes far more common non-synonymous changes. I would expect these to be an order of magnitude higher. I would imagine this data is available somewhere, but I couldn’t see it in CoV-GLUE. Most likely it can be extracted from GISAID which seems to be the data source used for CoV-GLUE. If someone would like to work on an analysis of non-synonymous mutations, please get in touch.
Also, I’d warn again drawing any strong conclusions from the analysis presented here. This is very much a first look at the data and an attempt to feel out the issue. I think it would be interesting to replicate/build out this work however, and would love to hear any comments.
Tarball of the (bad) code used here: Analysis.tar.gz
References/Notes
[1] https://www.cdc.gov/coronavirus/2019-ncov/downloads/rt-pcr-panel-primer-probes.pdf specifically the primers are:
GACCCCAAAATCAGCGAAAT
TCTGGTTACTGCCAGTTGAATCTG
TTACAAACATTGGCCGCAAA
GCGCGACATTCCGAAGAA
[2] https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2797725/
[3] cov-glue.cvr.gla.ac.uk
[4] Some messy scripts were required: http://41j.com/blog/2020/06/scripts-to-download-sars-cov-2-replacements/
[5] I used some awful awk to do this: for i in *; do awk ‘BEGIN{n=0;RS=”referenceNtCoord\”:\””;FS=”\”,\””;}{if(n==1) print $1;n++;}’ $i;done > mutlocs
[6] These are stored in the file called “primers”, in the tarball at the end of this post.
[7] This does a brute force alignment, looking for exact matches only on the forward and reverse strand. SARS-CoV-2 is only ~30Kb so computationally this is no problem.
[8] Within 5 bases of the 3′ end.