QuantumSi’s Protein Sequencing Approach
I’ve previously written about QuantumSi’s DNA sequencing work. Recently, QuantumSi have been promoting their protein sequencing platform. This may be a device that incorporates both DNA and protein sequencing functionality [1]. In this post I’m going to take a look at one of their protein sequencing patents [0] and review the approach.
Expectation Management
Before we dig into the technical details, let’s review the approach at a high level, in the context of DNA sequencing.
The basic process used to sequence proteins can be briefly described as follows:
- Isolate single proteins
- Attach a label to the terminal amino acid, and detect the label.
- Remove a single terminal amino acid.
- Go to step 2 to identify the next amino acid.
At a high level is not unlike single molecule sequencing-by-synthesis, in that monomers are detected sequentially. The difference here being that rather than incorporating monomers, in this approach they are cleaved.
While the basic process is similar to DNA sequencing, building the machinery to sequence proteins is far more complex. In DNA sequencing we have a bunch of tool (proteins) developed by nature which we can harness to develop sequencing approaches.
DNA’s complementary nature provides a simple approach to both amplifying polymers and introducing labels. We have a vast array of proteins that incorporate nucleotides, degrade nucleotides, and modify DNA sequences. For the most part, none of this machinery exists when working with proteins.
As we can’t amplify proteins, we are stuck with single molecule approaches. From DNA sequencing, we’ve seen that this alone limits our accuracy, and in general single molecule approaches have an error rate of >10% whereas amplified approaches (Illumina) have error rates significantly less than 1%.
Not only this, but the “alphabet” of proteins is an order of magnitude greater than for DNA sequencing. Beyond the ~20 standard amino acids, number of modified variants also exist further complicating labeling and identification.
Our base line expectation is therefore that initial data quality will be worse than DNA sequencing. This may not matter if the applications are compelling, as there’s not as much competition in the protein sequencing space.
Technical Approach
There are two technical approaches described in detail in the patent [2]. Both approaches use single molecule optical detection of a protein under sequencing attached to a surface (one example shows 18% occupancy [3]). The readout system appears to be similar to that mentioned in my previous post.
Sequencing Approach 1 – Label + Cleavage Enzyme
The first approach uses a labeled recognition enzyme. In this approach a (fluorescently) labeled recognition protein is used to detect the terminal base. From what I can tell these proteins don’t bind strongly, so they are transiently binding on and off.
At the same time, a cleavage enzyme is in the mix. The cleavage enzyme, at some appropriately low concentration comes in and removes terminal amino acids.
Example 6 shows what appears to be experimental data for this process. The example uses ATTO 542 label ClpS2. and an aminopeptidase (VPr). The protein they are attempting is sequence is YAAWAAFADDDWK.
ClipS2 binds to Y,W and F and apparently doesn’t bind to other terminal amino acids.
Two raw traces for this sequencing experiment are shown:
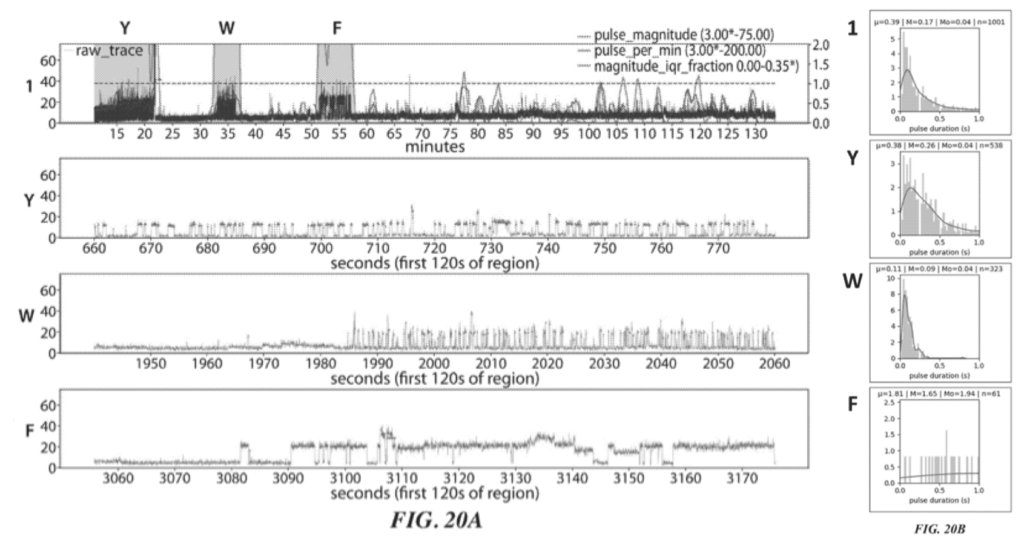
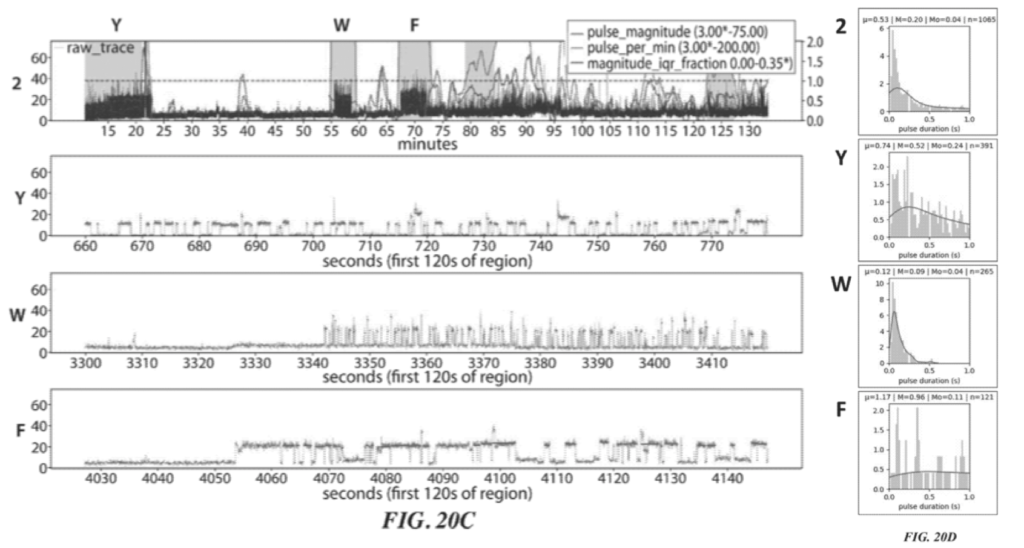
These two figures (20A and 20C) show two independent sequencing runs. Transient binding of ClpS2 to Y, W and F is shown. The experiment starts with the Y exposed as the terminal animo acid. As such we see transient binding of ClpS2 to Y from time point zero. At some point the cleavage enzyme comes in and the “Y” gets chopped off. “A” is now exposed at the terminal animo acid. ClpS2 shows no binding to “A” and we therefore don’t see any binding.
“A” then gets cleaved, exposing yet another “A” (still no binding). This is cleaved revealing W as the terminal base and we see transient binding again etc.
As it goes this is interesting, but it doesn’t really tell us anything about the sequence, which will naturally be runs of “F or W or Y” and not “F or W or Y”. It’s clear from the traces that the length of the transient binding period provides little informative information. For example, the time taken to cleave two “A” differs by a factor of 5 between the two experiments.
So in order to determine which of F, W, and Y was detected we need to look more closely at the transient binding. Figures 20B and 20D show histograms of the pulse duration. The variation and average durations seem to differ significantly for each animo acid.
This provides a basic proof of concept for animo acid detection. But it’s quite limited. The issues are as follows:
- There’s no mechanism for detecting runs of identical amino acids.
- The demonstration shows detection of 4 out of > 20 amino acids.
- While the durations are somewhat distinct for these amino acids, it seems likely that 20 such distributions would show significant overlap.
- The sequence is quite short (so we don’t know how long we can go without damaging the protein/other issues occurring).
Table 1 of the patent lists 33 amino acid recognition proteins, with 13 different binding patterns. These appear to cover 16 of the >20 amino acids ambiguously. A combination of there recognition proteins (and ideally a few more) might bring you closer to a full sequencing approach.
But, overall this work seems to provide a basic proof of concept of the detection process. With some additional work I could see this working as a protein fingerprinting technology. Where an ambiguous protein sequence is compared against a database of known protein sequences. In the above example the fingerprint might be something like:
“One or more Y”,”One or more not F,W,Y”,”One or more W”, “One or more not F,W,Y”, “One or more F”
With a long enough sequence, this may unambiguously identify a particular protein/class of proteins. However single point mutations might be more challenging.
Using additional recognition proteins would improve this fingerprinting process. Progressing this toward a sequencing platform seems challenging, and might require development of the technical approach.
Sequencing Approach 2 – Labeled Amino acid specific cleavage enzyme
Some data is also shown for a second approach. Here the cleavage enzyme is specific to certain animo acids. The demonstration shows that they have a method for incorporating labels into these exopeptides [5]. But there doesn’t seem to be any data demonstrating sequencing using this approach.
A list of amino acid specific exopeptides is provided [6]. However these seem to be far more limited than the recognition enzymes in approach 1. Only three types of specific exopeptides are listed those specific to Glu/Asp, Met or Proline.
This approach therefore seems more challenging and less developed.
Summary
Overall this seems like an interesting approach to a problem that hasn’t received that much attention. I’d expect the initial platform to be nearer to a “protein fingerprinting technology” than a full protein sequencing instrument. This seems like an interesting tool in its own right. If the initial instrument is framed as a protein sequencing platform, I would expect the error rate to be far higher than we’re used to seeing for DNA sequencing (probably in the order of >20%).
However, all this speculation is based on a single patent. They may have developed beyond this patent and it will be interesting to see what is finally released.
Notes
That’s it, you can stop reading now…
Ok, well… this section contains a few other notes from the patent. It doesn’t really add much to the discussion above, but they are here in part for my own reference. The footnotes below also support some of the assertions in the text above and may be of interest.
As is often the case, this patent mentions a number of other approaches which could be used. The patent discuss nanopore readout briefly. Indicating that the protein being sequenced could be immobilized on a nanopore. Recognition molecules (labels) are then detected through changes in conductance of the nanopore.
Other sections refer to “conductivity labels”.
Shielding elements. This is essentially a protein (or other element) that shields the recognition molecule from photo damage. Shield proteins are used in other single molecule sequencing approaches, and a number of methods are presented in the patent.
Other approaches to removing terminal amino acids are mentioned… Edman degradation, phenyl isothiocyanate.
Various other detection methods are briefly discussed for example Aptamers.
While I’ve not mentioned it in the text above there are some other nice plots of the pulse duration differences for ClpS2 in example 5:
Example 5: ClpS2 as recognition label. ClpS2 is labeled with dye. Single molecule intensity traces shown in figure 19B.
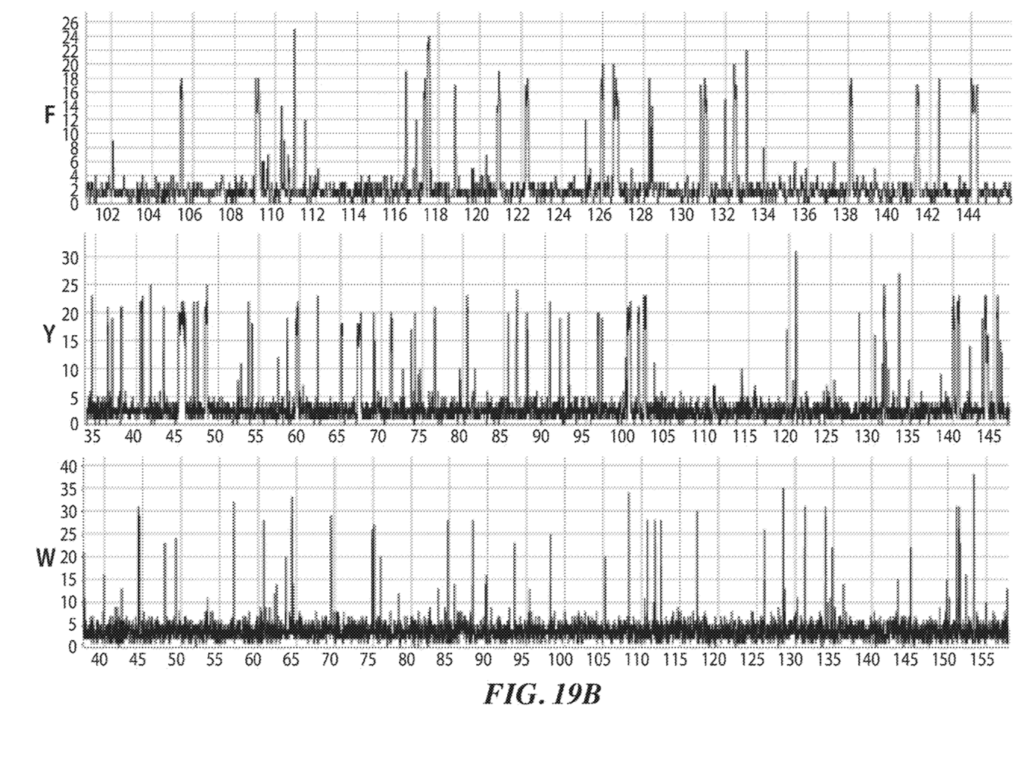
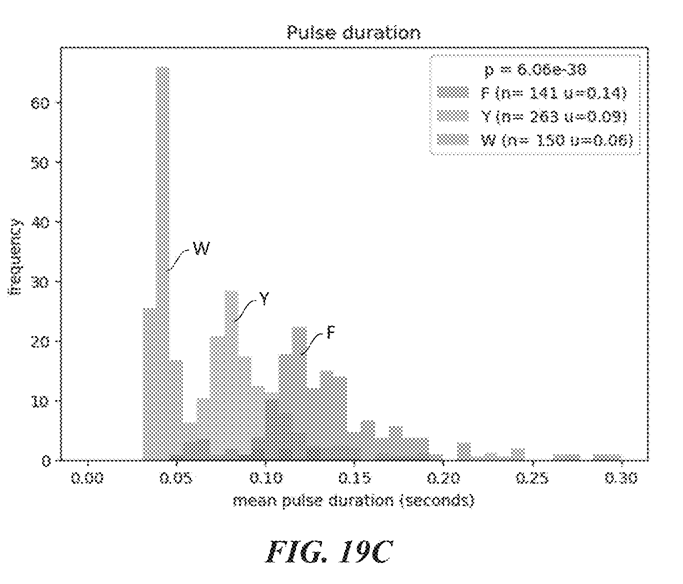
Similar plots are shown for ClpS, and ClpS1.
Footnotes
[0] https://www.freepatentsonline.com/y2020/0209257.html US20200209257A1
[1] “In some aspects, the application relates to the discovery of polypeptide sequencing techniques that allow both genomic and proteomic analyses to be performed using the same sequencing instrument.”
“Such strategies may require modification of an existing analytic instrument, such as a nucleic acid sequencing instrument, which may not be equipped with a flow cell or similar apparatus capable of reagent cycling. The inventors have recognized and appreciated that certain polypeptide sequencing techniques of the application do not require iterative reagent cycling, thereby permitting the use of existing instruments without significant modifications which might increase instrument size.”
[2] As always, a number of other approaches are also mentioned. But these are the approaches that have the best supporting data. I review some of the alternative approaches at the end of the post.
[3] One example shows proteins attached using a DNA linker, with 18% single protein occupancy. This seems lower than Poisson. Only single wells will be sequencable so this limits throughput. Example 2 (page 127).
[4] Amino acid recognition proteins. Table 1. Lists 33 amino acid recognition proteins and their preferred binding. Many of these appear to prefer the same amino acids. There are therefore 13 different types of “preferred binding” listed: FWY: 4, FWYL: 6, FWYLVI: 1, phosphorus-Y: 5, FWYLI: 1, KR: 1, DE: 1, KRH: 3, P: 2, KRHWFY: 5, PMV: 1, G: 2, A: 1.
[5] They reference the following paper, which uses non-natural amino acids. Chin J. W et al. J Am Chem Soc. 2002 Aug. 7 124(31):9026-9027
[6] Aminopeptidases. Table 3 lists amino peptidases. These should selectively cleave terminal amino acids. There seem to be 3 classes here with limited coverage of the amino acid space (Glu/Asp: 1, Met: 2, Proline: 6). Table 4 provides a much longer list of non-specific Amino-peptidases.