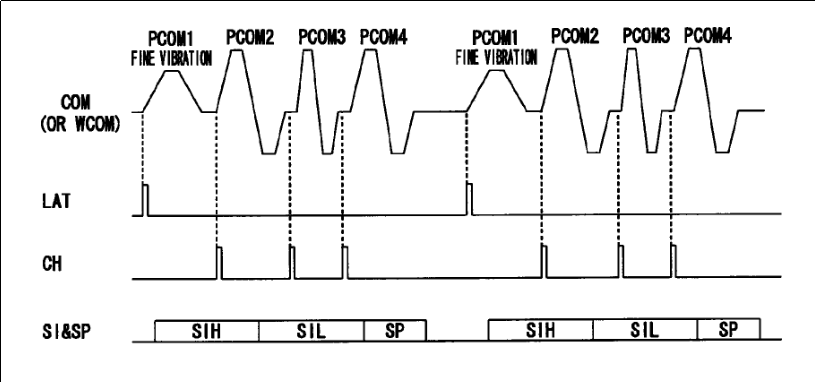
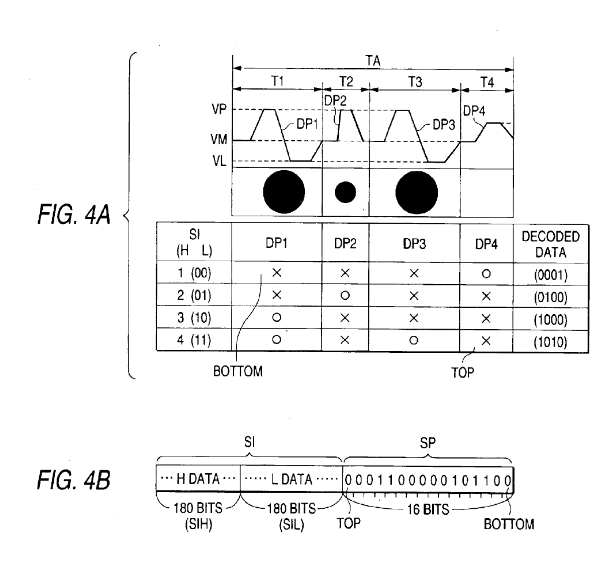
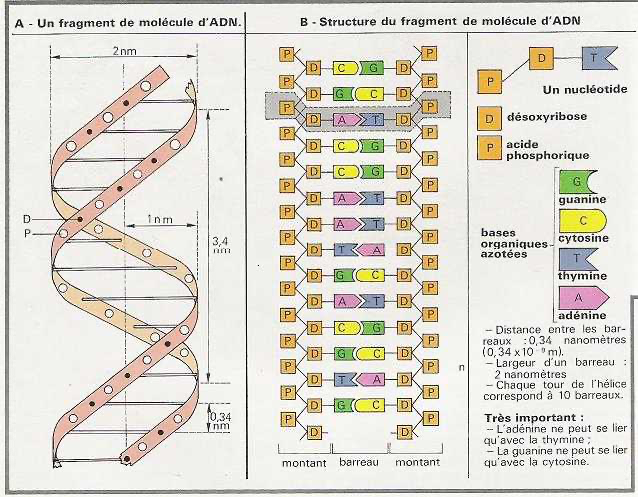
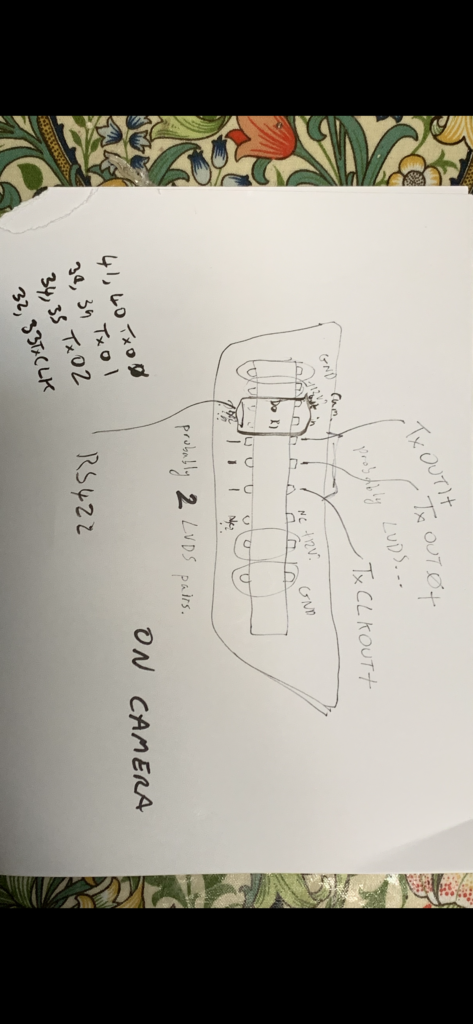
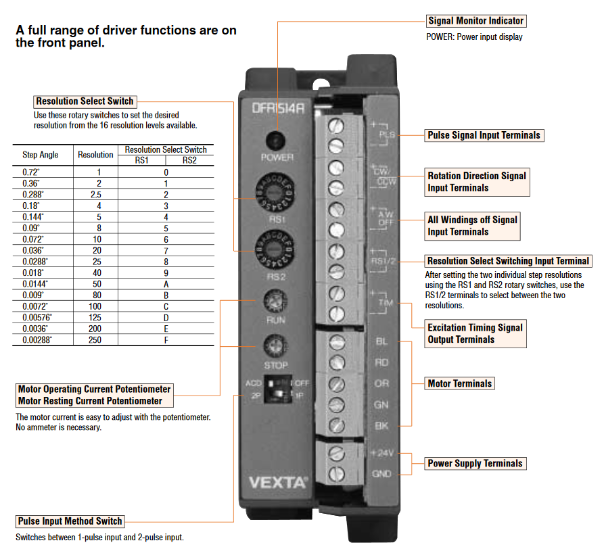
SCK : Clock for SI&SP pin 13 LAT : Latch signal SI&SP: Drive pulse selection data, selects nozzle COM : Drive signal CH : Connect drive input to nozzle
Single molecule diffusion on hard, soft and fluid surfacesRhodamine 6G (R6G) and Rhodamine 123 (R123), both of which can readily absorb on the three selected model surfaces by hydrophobic interaction.octadecyl-triethoxylsilane (OTE)
Direct Measurement of Single-Molecule Diffusion and Photodecomposition in Free SolutionIndividual rhodamine- 6G (R6G) and rhodamine-labeled 30-base single-stranded DNA (ssDNA) (DNA- R6GThe predicted full-width diffusion distances, for R6G and DNA-R6G at a 100-ms exposure time, are 75 pixels and 35 pixels (each pixel 200nm so 15 and 7micron).
Set up hot water bath at 55-60� C and an ice water bath.
For each onion, make a solution consisting of one tablespoon (10 ml) of liquid dishwashing detergent or shampoo and one level 1/4 teaspoon (1.5 g) of table salt. Put in a 1-cup measuring cup (250 ml beaker). Add distilled water to make a final volume of 100 ml. Dissolve the salt by stirring slowly to avoid foaming.
Coarsely chop one large onion with a knife and put into a 4-cup measuring cup (1000 ml). For best results, do not chop the onion too finely. The size of the pieces should be like those used in making spaghetti. It is better to have the pieces too large than too small.
Cover chopped onion with the 100 ml of solution from step 2. The detergent dissolves the fatty molecules that hold the cell membranes together, which releases the DNA into the solution. The detergent, combined with the heat treatment used in step 5, causes lipids (fatty molecules) and proteins to precipitate out of the solution, leaving the DNA. The salt enables the DNA strands to come together.
Put the measuring cup in a hot water bath at 55-60� C for 10-12 minutes. During this time, press the chopped onion mixture against the side of the measuring cup with the back of the spoon. Do not keep the mixture in the hot water bath for more than 15 minutes because the DNA will begin to break down.
Cool the mixture in an ice water bath for 5 minutes. During this time, press the chopped onion mixture against the side of the measuring cup with the back of the spoon. This step slows the breakdown of DNA.
Filter the mixture through a #6 coffee filter or four layers of cheese cloth placed in a strainer over a 4-cup measuring cup. When pouring the mixture into the strainer, avoid letting foam get into the measuring cup. It can take more than an hour to recover most of the liquid. The filtering can be done in a refrigerator overnight.
Dispense the onion solution into test tubes, one for each student. The test tube should contain about 1 teaspoon of solution or be about 1/3 full, whichever is less. For most uniform results among test tubes, stir the solution frequently when dispensing it into the tubes. The solution can be stored in a refrigerator for about a day before it is poured into the test tubes. When the solution is removed from the refrigerator, it should be gently mixed before the test tubes are filled.
The process of extracting DNA from a cell is the first step for many laboratory procedures in biotechnology. The scientist must be able to separate DNA from the unwanted substances of the cell gently enough so that the DNA is not broken up.
Your teacher has already prepared a solution for you, made of onion treated with salt, distilled water and dishwashing detergent or shampoo. An onion is used because it has a low starch content, which allows the DNA to be seen clearly. The salt shields the negative phosphate ends of DNA, which allows the ends to come closer so the DNA can precipitate out of a cold alcohol solution. The detergent causes the cell membrane to break down by dissolving the lipids and proteins of the cell and disrupting the bonds that hold the cell membrane together. The detergent then forms complexes with these lipids and proteins, causing them to precipitate out of solution.
PROCEDURE
Add cold alcohol to the test tube to create an alcohol layer on top of about 1 cm. For best results, the alcohol should be as cold as possible. The alcohol can be added to the solution in at least three ways. (a) Fill a pasteur pipette with alcohol, put it to bottom of the test tube, and release the alcohol. (b) Put about 1 cm of alcohol into the bottom of a test tube and add the onion solution. (c) Slowly pour the alcohol down the inside of the test tube with a pasteur pipette or medicine dropper. DNA is not soluble in alcohol. When alcohol is added to the mixture, all the components of the mixture, except for DNA, stay in solution while the DNA precipitates out into the alcohol layer.
Let the solution sit for 2-3 minutes without disturbing it. It is important not to shake the test tube. You can watch the white DNA precipitate out into the alcohol layer. When good results are obtained, there will be enough DNA to spool on to a glass rod, a pasteur pipette that has been heated at the tip to form a hook, or similar device. DNA has the appearance of white mucus.
“a pressure source which provides a gas load pressure valve to the reservoir chamber which is sufficiently negative such that fluid adjacent and facing the orifice is loaded into the head by being drawn into the reservoir chamber through the orifice and delivery chamber, while simultaneously being insufficient to result in ambient atmosphere entering the delivery chamber through the orifice once the head has been loaded and no further fluid is facing and adjacent the orifice. “: https://patents.google.com/patent/US6323043B1/en
“I never saw about 10% of my paycheck. Turned out that the person working in HR was taking it for herself. The finance department apparently knew that this was going on. I can only imagine the other types of fraudulent things going on in this company amongst others. The management obviously don’t care about the employees at all, especially after the recent mass layoff. Apparently only around 5 people are left after the last mass layoff, taking whatever funds are left for themselves before they retire or if they ever sell whatever’s left of this sad excuse of a company. Outlook for that isn’t so great though, since the market for cataract surgery devices is so small and is already being outcompeted by larger companies with better talent and technologies like ORA. Honestly, their investors are just sinking dead cash into a lost cause at this point. I hope they finally pull the plug.”
GigE Vision, Sony ICX274 EXview CCD sensor, auto iris, 25 fpsEdit
Prosilica GC1600H is a 2.0 Megapixel camera with a GigE Vision compliant Gigabit Ethernet interface and Hirose port. Prosilica GC1600H is offered in both monochrome and color models. This camera incorporates the high quality Type 1/1.8 (8.923 mm diagonal) Sony ICX274 CCD sensor with Super HAD CCD technology that provides superior image quality, excellent sensitivity, and low noise. At full resolution, this camera has a frame rate of 25 frames per second. With a smaller region of interest higher frame rates are possible. By default monochrome models ship with no optical filter and color models ship with an IRC30 IR cut filter.
Benefits and features:
Monochrome (GC1600H) and color (GC1600CH) models
GigE Vision interface
Screw mount RJ45 Ethernet connector for secure operation in industrial environments
Supports cable lengths up to 100 meters (CAT-5e or CAT-6)
Popular C-Mount lens mount
Easy camera mounting via standard M3 threads or optional tripod adapterr
Responds to I2C commands using this pinout as reference, board supplied with 5v, using 3.3v external pullups. Module contains required 2.8v and 1.8v regulators. One other unidentified regulator? Appears to use parallel bus.
I2C responds on address 0x49. This is not the correct device ID for MT9P. Commands do not appear to match MT9P:
e.g. device ID:
I2C>[0x92 0 NACK I2C START BIT WRITE: 0x92 ACK WRITE: 0x00 ACK I2C>[0x93 r r I2C START BIT WRITE: 0x93 ACK READ: 0x17 READ: ACK 0xB0
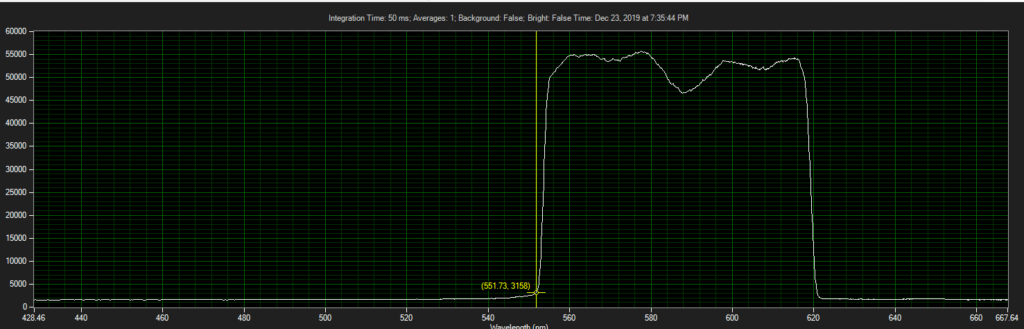
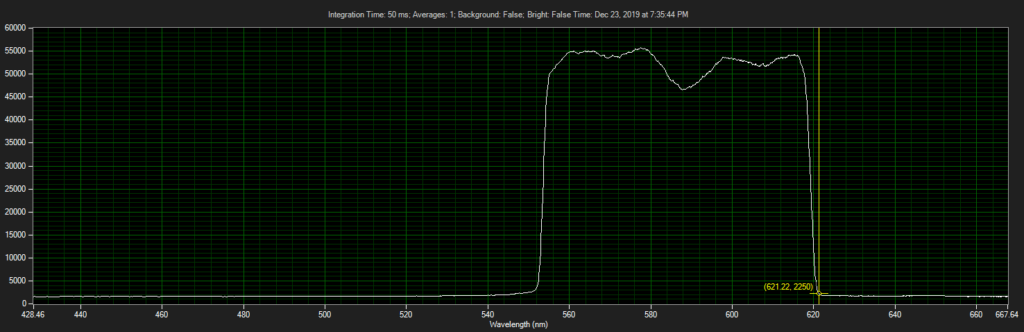
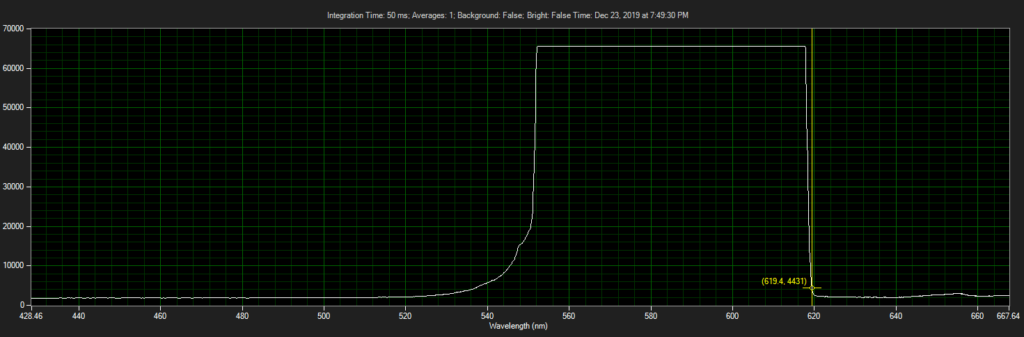
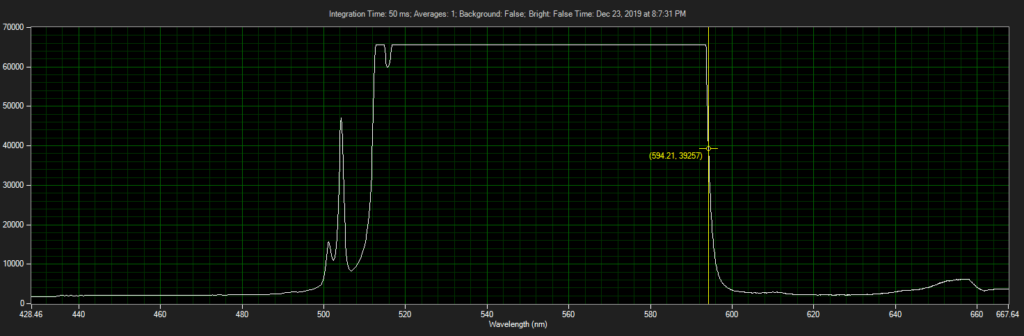
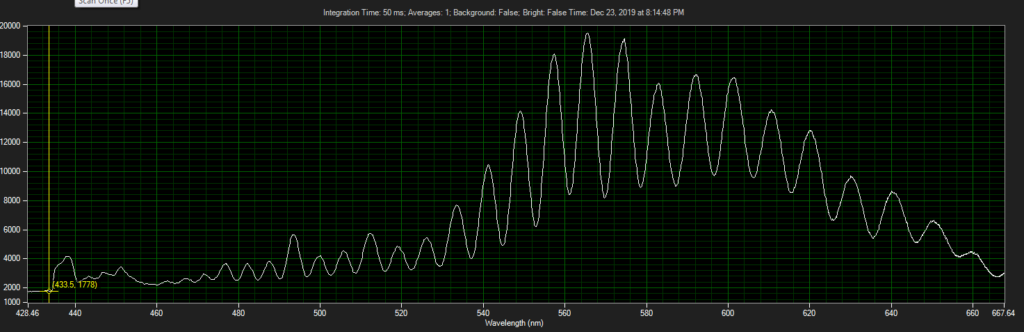
CCD has filters which pass ~550 to 620.
Spectrum of filter on CCD, from Xenon light source:
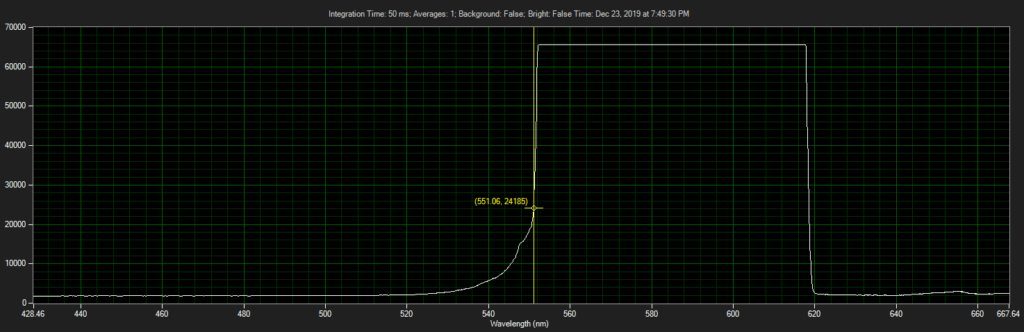
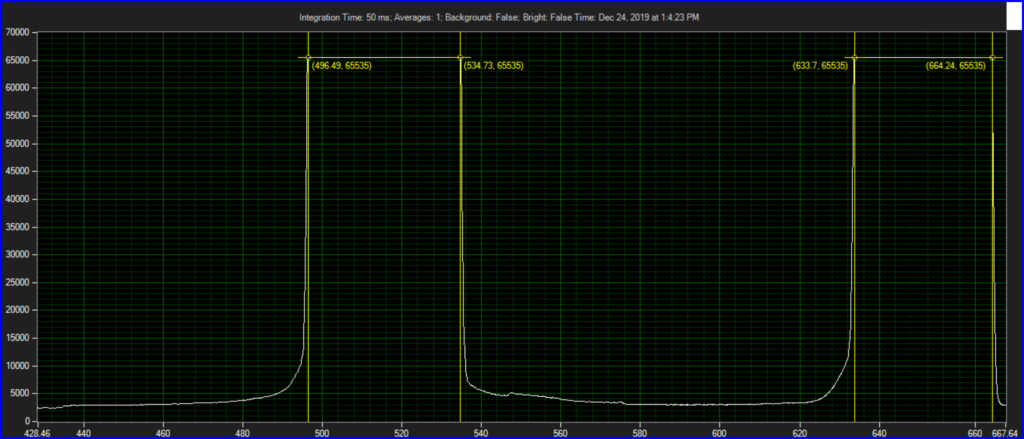
Filter on CCD optics, nearer objective:
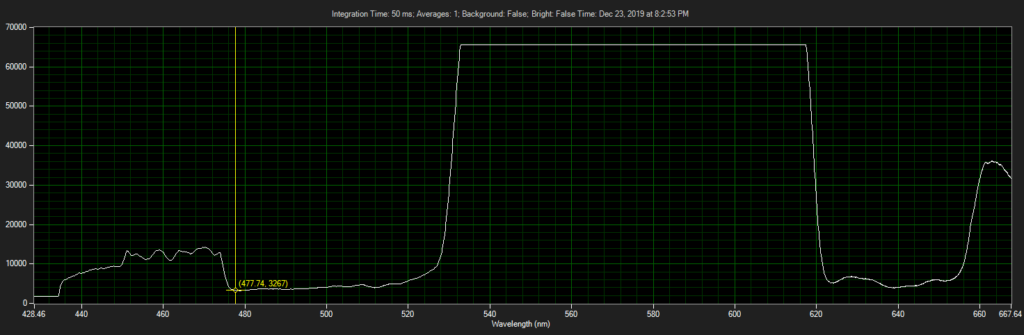
Mirror on objective. Called filter 3 on ebay, reflect toward CCD of Xenon:
I picked this up at auction cheap because it looked entertaining. It’s labeled as an “Infrared Microscope” but essentially as far as I can tell it’s general used as an accessory to FT-IR 6000 series IR spectrometers.
The spectrometers have a built in detector, but can be configured to take IR light routed through the microscope. So you use the microscope built in illumination to locate the part of the sample you’re interested in on a slide. Then you can route the IR input/output through the spectrometer and possibly (I’m not entirely certain) use the detector built into the microscope while using the interferometer components of the spectrometer.
My name is Nava Whiteford. I’ve worked for a few sequencing companies. I have equity in a few sequencing companies based on my previous employment (I try to be unbiased in my posts). You can contact me at: [email protected]