I was trying to interface directly with the I2C on the Arducam Pivistation 5 Klarity camera module for… reasons.
Anyway I figured this would just expose the I2C on the IMX283… but it doesn’t seem to. There seems in fact to be another little microcontroller (GD e230f8?) there which uses a different I2C command set. It seems similar to the interface from this camera:
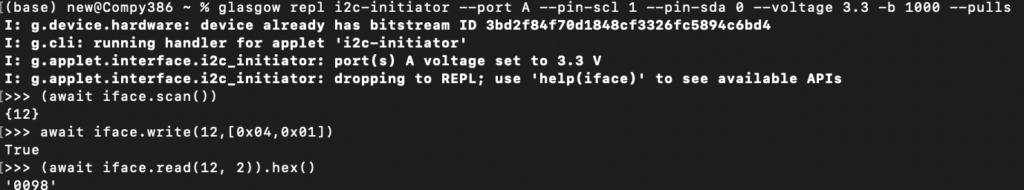
Anyway… I was able to get some info out of it using a Glasgow Interface Explorer. This note is here so someone else doesn’t waste time wondering why the IMX283 (which I think should respond on 0x1a) is responding on 0x0c on this camera.
The Glasgow commands required are below:
Also, it seems that you can crash the microcontroller pretty easily by doing additional reads?
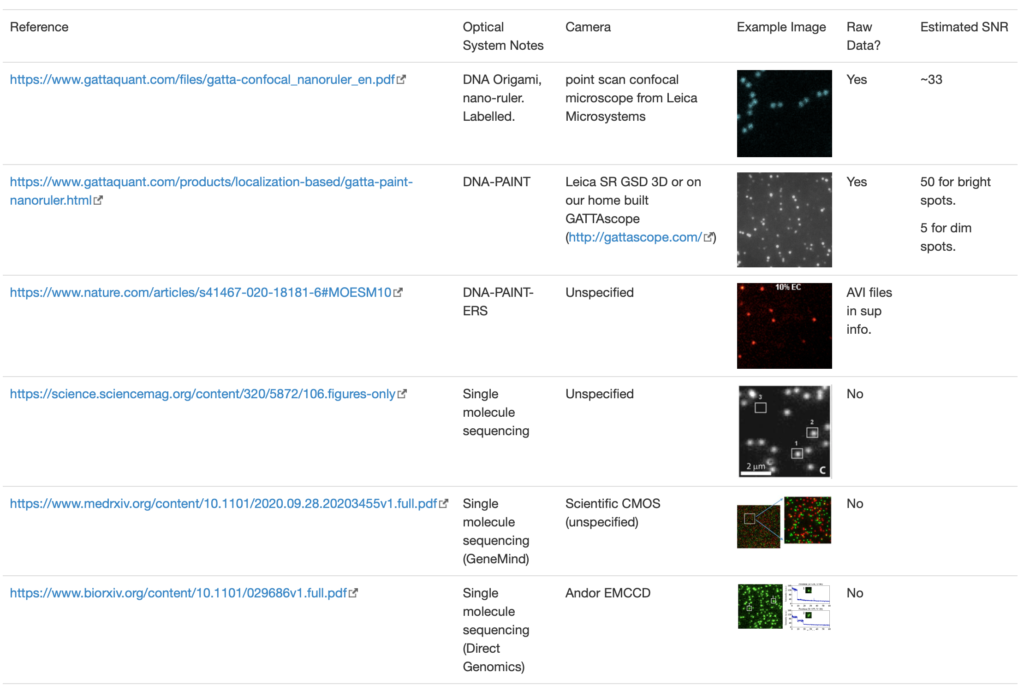
“If we recall that an Ampere corresponds to a charge flow of 1 Coulomb each second and further, use the fact that the charge on a monovalent ion is approximately 1.6 x 10-19 Coulombs (that is 1 electron or 1 proton charge), then we see that a current of about one pA corresponds to roughly 107 ions passing through the channel each second. This value is in agreement with measurements” http://book.bionumbers.org/how-many-ions-pass-through-an-ion-channel-per-second/
Double stranded Lambda DNA (16um long, 48kb). Translocates in 3 different confirmations. Grabbed at the end. Grabbed in the middle (completely folded), Grabbed near an edge (partly folded). Difference in current levels is ~1nS between each confirmation (200pA). 22nm pore is used. Translocation time ~3ms.
My name is Nava Whiteford. I’ve worked for a few sequencing companies. I have equity in a few sequencing companies based on my previous employment (I try to be unbiased in my posts). You can contact me at: [email protected]