Evonetix and other thoughts
This post isn’t really about the Evonetix approach to DNA synthesis, but rather about related ideas. To set the context, I review an approach described in one of their older patents (their current approach pretty well on their website).
Evonetix was incorporated in February 2015 (Cambridge, UK). They have raised somewhere of the order of ~15MUSD to date. Investors include Cambridge Consultants, Hermann Hauser, DCVC, Draper Esprit, Morningside Group, Rising Tide, and Civilization Ventures.
Their patents [1] describe the basic approach, their patents seem quite readable and I recommend taking a look.
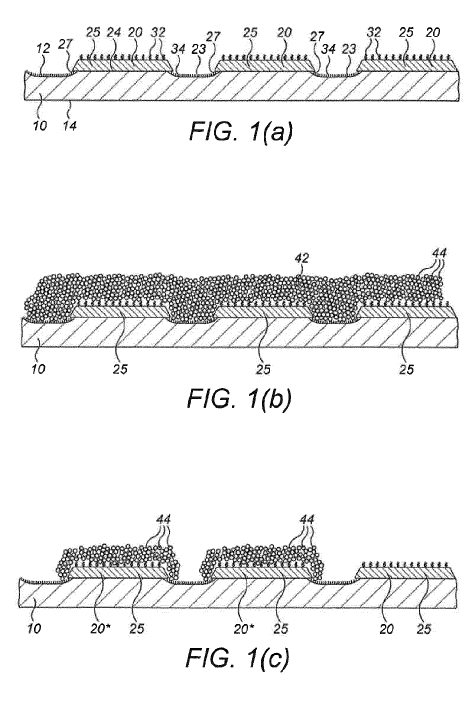
Essentially, they describe using a substrate on which sites are coated with a waxy layer (it seems n-Alkane is preferred). They then selectively melt the wax in order to expose the substrate to reagents. They don’t mentioned enzymatic DNA synthesis methods in the patents I’ve read at all. It looks like phosphoramidite synthesis is the focus.
They discuss two approaches to applying heat, either using a laser or via on chip heating elements. Given their recent deal with LioniX [4] it seems likely that they will continue down the silicon based route. As usual a number of configurations are discussed in the patent, but in particular a 0.5 micron spacing is mentioned. In the patent and elsewhere a billion wells seems to be the target. This would result in something like a ~16mm^2 chip. Big enough that I’d hope it would be reusable, but not massive.
I would guess one of the issues would be insuring that the thermal changes are sufficiently localized. The patent suggests a couple of methods [2] [3] to help with this, but I imagine it will be a significant challenge. In fact, you also want to uniformly cool the chip after each cycle as well… so all round accurate thermal control will be the important.
Thoughts
Essentially the Evonetix play is around designing a system to selectively expose a “virtual well” to reagents. That’s quite an interesting idea, but it made me wonder about related approaches.
Activating Enzymes
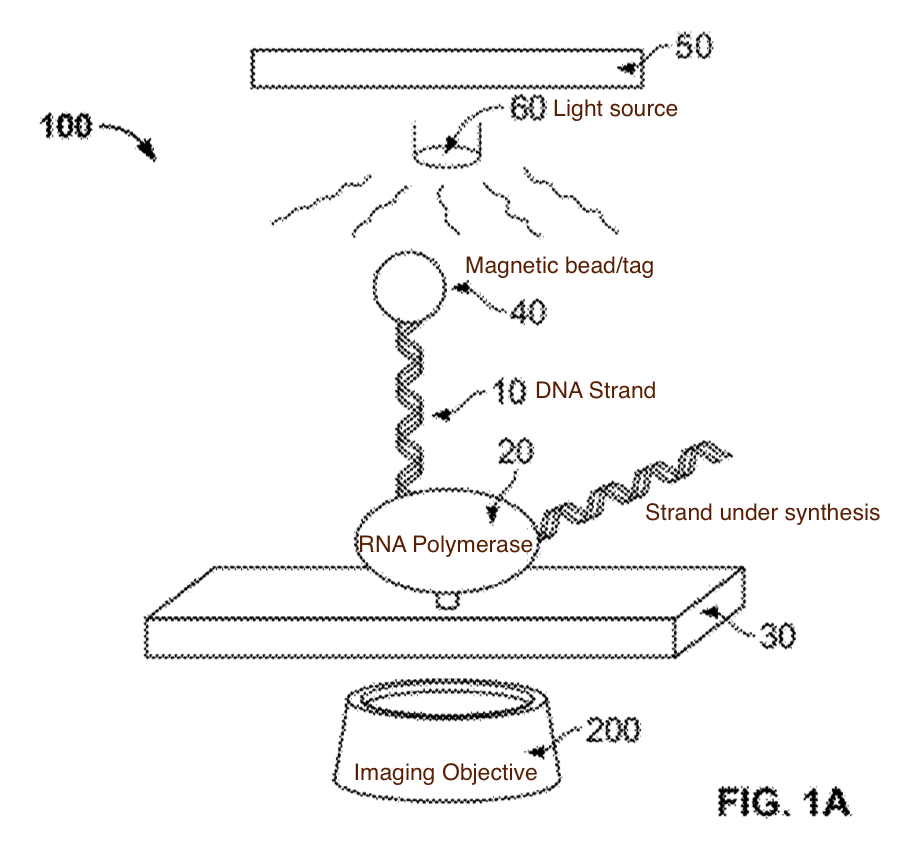
The first thought that occurs to me is that how might a similar system be used in an enzymatic DNA synthesis platform. Rather than using heating elements to expose a strand under extension to reagents could you use heat to either activate or deactivate enzymes.
For example lets say you cool the whole chip down, to a point where the template independent polymerase (TdT) is largely inactive. You then locally heat the chip, only where you want incorporation to take place (which would occur cyclicly).
Confining the heat in the presence of reagents might be more of an issue here.
Using an electric field
Rather than deactivating the polymerase thermally, could you alter the local conditions around a strand under synthesis using an electric field. Initially I was wondering if a negative field would, kind of push away nucleosides. However, it seems [5] that a field can also be used to locally change the pH. Could you therefore use local changes in pH to activate/deactivate an enzyme?
Non-virtual wells
The Evonetix play is around the use of a “virtual well”. But how might you go about creating real wells with caps which could easily be opened and closed to selectively expose the contents of the well to reagents?
One approach might be to use a larger (maybe 500nm?) bead as a cap. The bead could be magnetic, or charged. In those cases, you could use a magnetic or electric field to selectively push the bead away from the well just enough to allow reagents to enter the well. You might want to coat the well with something to help create a seal perhaps.
Local heating could also be used to push the bead off the well perhaps, or other methods like optical tweezers… these seem less attractive however.
Those were my initial thoughts anyway… Evonetix seem like an interesting company, and I look forward to seeing how things develop.
Notes
[1] https://patents.google.com/patent/US20160184788A1/en US20160184788A1
[2] “To achieve a great melting and coalescence of the masking material within a limited area, a series of discrete heating pulses may be used, each pulse being separated by period of cooling. For example, a series of 1,000-20,000…heating pulses… about 1 ns… with about 1 microsecond of cooling between each pulse may be suitable.”
[3] “The thermal energy may be applied to the selected ones of the sites simultaneously, but alternatively it may be desired to stagger the application of thermal energy to the select sites in order to allow for more efficient diffusion of thermal energy away from a selected site following the application of thermal energy. Suitably, the application of thermal energy to the selected site is carried out so that the adjacent sites are not heated simultaneously and optionally not immediately one after another.”
[4] https://www.evonetix.com/evonetix-lionix/
[5] https://patents.google.com/patent/US20130344539