It’s been a while since we heard much from Direct Genomics. This Shenzhen based sequencing company was a reboot of early NGS player and Quake spinout Helicos.
The company is also notable for being founded by Jiankui He. He is the scientist responsible for the germ-line genetic editing of 3 human babies. And is reportedly serving a 3 year jail sentence.
One of the last papers referring to the Direct Genomics sequencing platform is this one from 2017. Where they discuss the Direct Genomics GenoCare single molecule sequencing platform.
With He’s jail sentence the future of Direct Genomics seemed uncertain. But it looks like the GenoCare platform still exists. Now under development by GeneMind, a Shenzhen based Biotech company founded in 2012. A couple of papers appearing this year describing the platform under their stewardship.
In particular a medRxiv paper from September this year, describes a new two color single molecule sequencing approach.
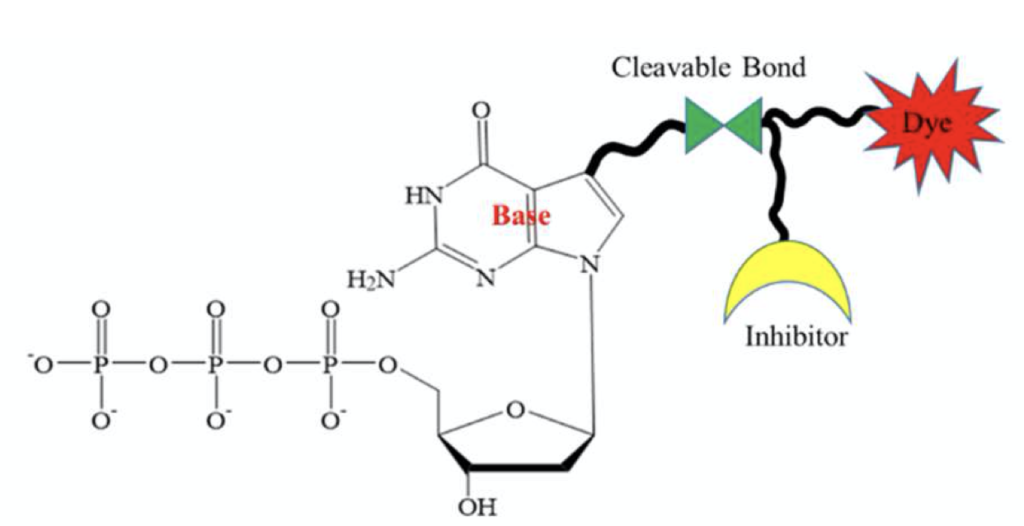
This seemingly uses the same virtual terminators as the original Helicos approach, with the dye and “terminator” sitting on the base:
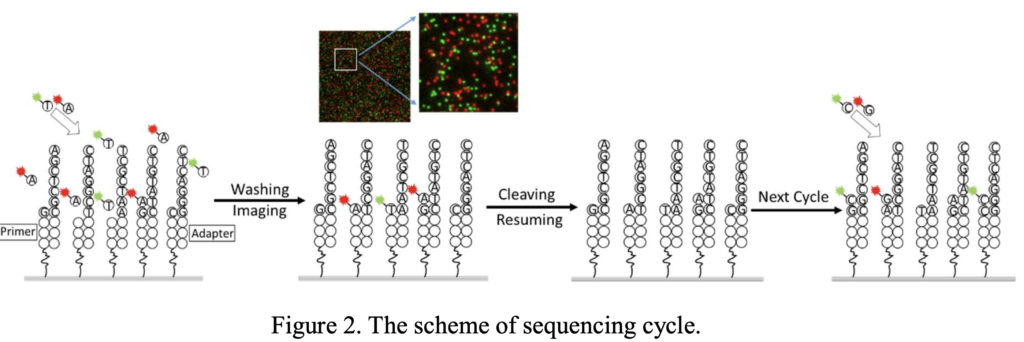
GenoCare two color sequencing isn’t the same as the Illumina two color approach, and doesn’t appear to provide the same advantages. On GenoCare’s platform two terminators (C and T) are labelled with a green dye, and two others (G and A) with a red dye [1].
This means that of each cycle, 4 images still need to be acquired. One set after flowing in C and A. And another after cleaving the dyes and flowing in T and G [2]:
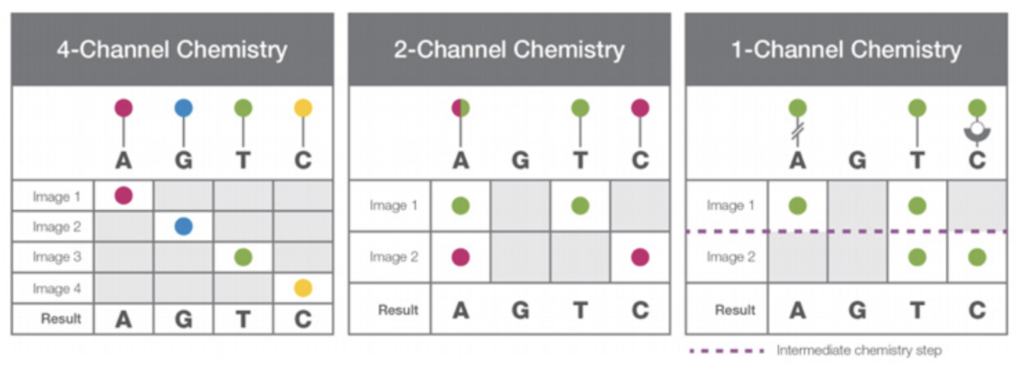
In contrast to this, Illumina two color sequencing incorporates information from “dark” bases. This means they can take only two images, and perform no intermediate chemistry in their two color approach:
Illumina’s 4,2 and 1 channel chemistries compared. Image from Illumina.
The GenoCare two color approach therefore doesn’t provide any advantage in terms of imaging speed/performance. Though the optics is likely cheaper than a four color system, it’s unclear as to why they don’t use a two color approach similar to Illumina’s which would allow them to take only two images, with no intermediate cleavage step on the same optical system. Perhaps there are IP issues here.
The GenoCare optical system uses objective style TIRF and a sCMOS camera. This appears to generate reasonable quality images, showing an SNR of >10. There have been huge advances in single molecule imaging since Helicos and this should simplify imaging/data analysis.
However, this doesn’t seem to have translated into massive improvements in data quality. With the GenoCare system showing mismatch, insertion, and deletion error rates of 0.61%, 1.45%, and 2.76% respectively.
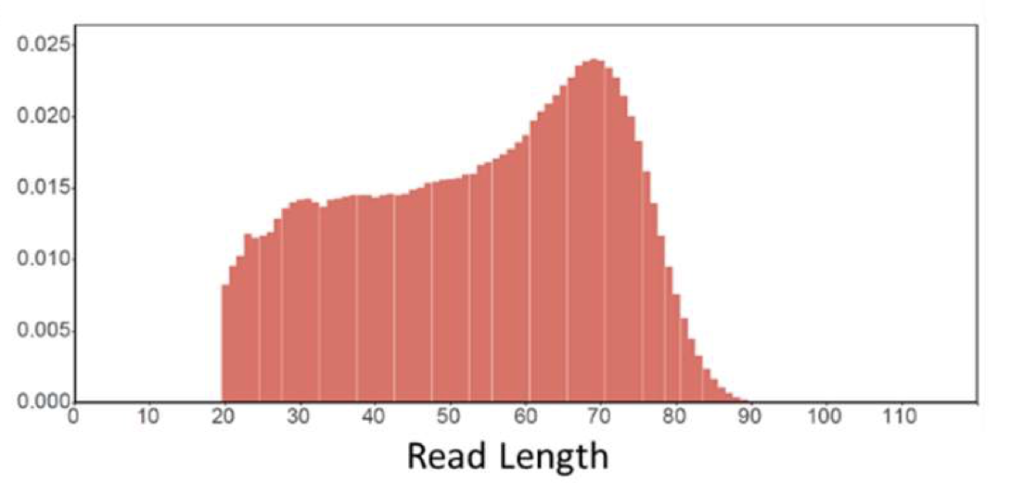
Reads are generally short, but usable. With a lopsided distribution heavy in short reads:
Overall, the GenoCare system has an error rate one to two orders of magnitude higher than the market dominating Illumina approach. Their optical system is likely more expensive (requiring single molecule sensitivity). And read lengths are shorter.
The sole advantage of this platform is that amplification is not required. This may simplify sample preparation, and result in lower bias for certain applications. Of course also the case for other single molecule approaches (PacBio, nanopore).
Overall, it’s difficult to see how this platform could be competitive with Illumina (or the largely similar SBS approach used by MGI)… but perhaps they’ll manage to find a way forward.
Notes
[1] “Two terminators are labeled with a green dye, whose peak fluorescent emission wavelength is 552 nm, while the other two terminators are labeled with a red dye with peak fluorescent emission at 664 nm.”. The paper doesn’t appear to not which dyes are associated with which bases, but this combination seems likely from the figures.
[2] The exact combination isn’t clear from the paper, but they can only image one red and one green labelled terminator per imaging cycle to generate an unambiguous read.
A couple of people have asked me about a new publication claiming a $15 genome. The paper seems to suggest that by reducing reagent costs, the cost of sequencing a genome can be dramatically reduced, by a factor of 10 to 100x.
To support this claim the paper suggests that: “SCT has a much thinner reagent layer and unlike flow chips and has no tubing which decreases reagent usage by orders of magnitude. Since the reagent cost accounts for 80-90% of current NGS platforms this decrease in reagent usage of SCT drops the operating costs from about $1000 to about $15 for WGS.”
The problem of course, is that the authors don’t have access to Illumina’s cost of goods. They just go by the cost Illumina sell their kits for. And as such you can’t use this in any way to justify that the reagent costs are 90% of the sequencing cost.
I’ve seen this a number of times from researchers. Who imagine that reducing the quantity of reagents used will have a dramatic effect on the cost of sequencing.
Moreover, there’s a lot more than just reagents in Illumina’s consumables costs. In addition to the raw costs of the reagents among other costs there’s at least: packaging, logistics, and the cost of a nano patterned flow cell.
It’s reasonable to imagine that this nano patterned flowcell is the bulk of Illumina’s consumable costs of goods. And as such using a smaller quantity of reagents likely has minimal effect on costs.
What I did like about the paper is that it provides a potential method for simplifying the fluidics system. A simple, compact sequencing platform which just has to flow a single buffer over the flow cell [2] seems like a neat idea. But I don’t expect it to massive impact costs.
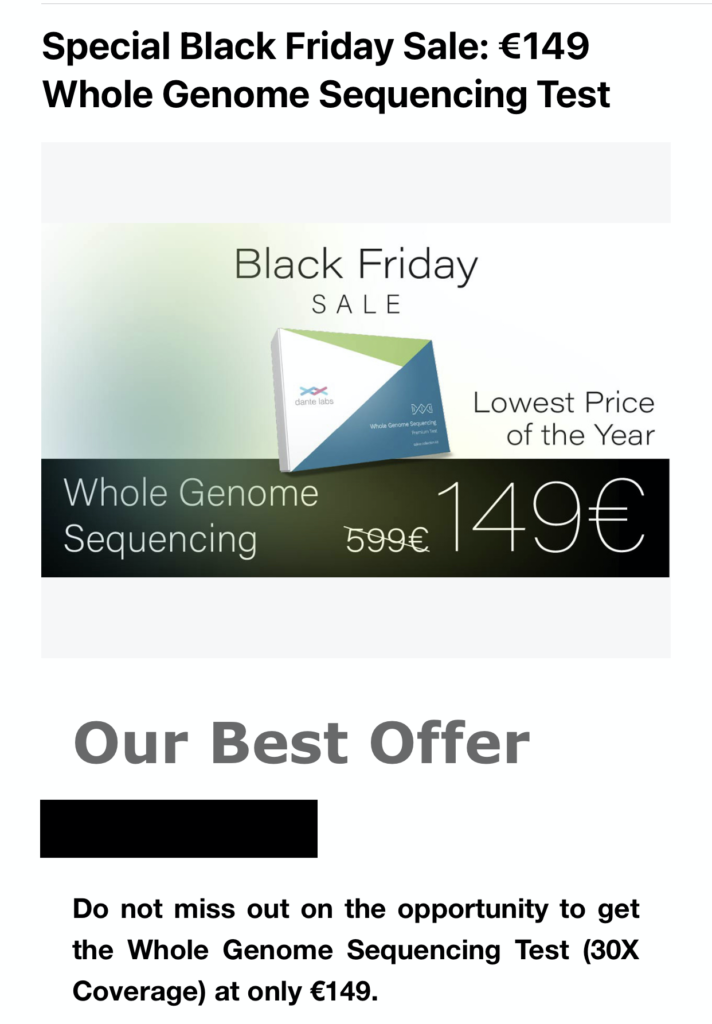
Talking of costs, Dante labs have recently been offering 30x human genomes for 150Euros:
Some have suggested that Dante sell their sequencing at a loss, and make up for it elsewhere… however I suspect the answer is that Dante uses MGI sequencing which they can get far cheaper than Illumina.
Dante don’t specify the technology used, but data quality metrics they’ve previously presented looked very similar to MGIs current offerings. Illumina have blocked the sale of MGI instruments in the US, and filed suits against them in the EU. This may in part explain why Dante are somewhat vague about who exactly is supplying their sequencing services.
Notes
[1] Some relevant quotes from this paper:
“When cost data were only available for a kit as a whole, kit costs were apportioned equally across all items in the kit.”
“The most expensive item was the HiSeq 4000 sequencing machine (Illumina), which cost £474,373, with an annual maintenance cost of £55,641. This sequencing system requires two consumable kits (a HiSeq 3000/4000 Sequencing by Synthesis [SBS] Kit costing £4207 and a HiSeq 3000/4000 Paired End [PE] Cluster Kit costing £2597), with half of each kit required per case.”
I’ve previously written about QuantumSi’s DNA sequencing work. Recently, QuantumSi have been promoting their protein sequencing platform. This may be a device that incorporates both DNA and protein sequencing functionality [1]. In this post I’m going to take a look at one of their protein sequencing patents [0] and review the approach.
Expectation Management
Before we dig into the technical details, let’s review the approach at a high level, in the context of DNA sequencing.
The basic process used to sequence proteins can be briefly described as follows:
Isolate single proteins
Attach a label to the terminal amino acid, and detect the label.
Remove a single terminal amino acid.
Go to step 2 to identify the next amino acid.
At a high level is not unlike single molecule sequencing-by-synthesis, in that monomers are detected sequentially. The difference here being that rather than incorporating monomers, in this approach they are cleaved.
While the basic process is similar to DNA sequencing, building the machinery to sequence proteins is far more complex. In DNA sequencing we have a bunch of tool (proteins) developed by nature which we can harness to develop sequencing approaches.
DNA’s complementary nature provides a simple approach to both amplifying polymers and introducing labels. We have a vast array of proteins that incorporate nucleotides, degrade nucleotides, and modify DNA sequences. For the most part, none of this machinery exists when working with proteins.
As we can’t amplify proteins, we are stuck with single molecule approaches. From DNA sequencing, we’ve seen that this alone limits our accuracy, and in general single molecule approaches have an error rate of >10% whereas amplified approaches (Illumina) have error rates significantly less than 1%.
Not only this, but the “alphabet” of proteins is an order of magnitude greater than for DNA sequencing. Beyond the ~20 standard amino acids, number of modified variants also exist further complicating labeling and identification.
Our base line expectation is therefore that initial data quality will be worse than DNA sequencing. This may not matter if the applications are compelling, as there’s not as much competition in the protein sequencing space.
Technical Approach
There are two technical approaches described in detail in the patent [2]. Both approaches use single molecule optical detection of a protein under sequencing attached to a surface (one example shows 18% occupancy [3]). The readout system appears to be similar to that mentioned in my previous post.
Sequencing Approach 1 – Label + Cleavage Enzyme
The first approach uses a labeled recognition enzyme. In this approach a (fluorescently) labeled recognition protein is used to detect the terminal base. From what I can tell these proteins don’t bind strongly, so they are transiently binding on and off.
At the same time, a cleavage enzyme is in the mix. The cleavage enzyme, at some appropriately low concentration comes in and removes terminal amino acids.
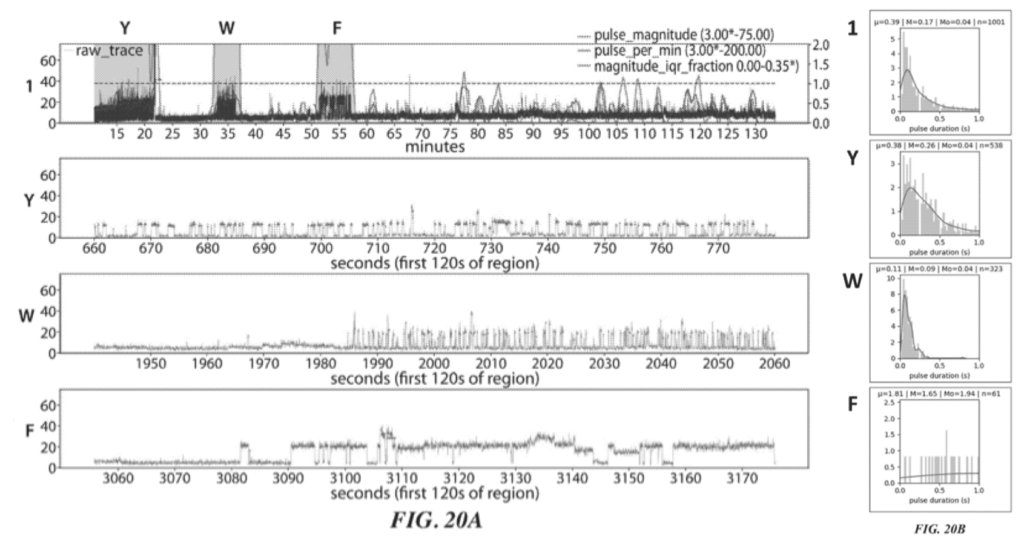
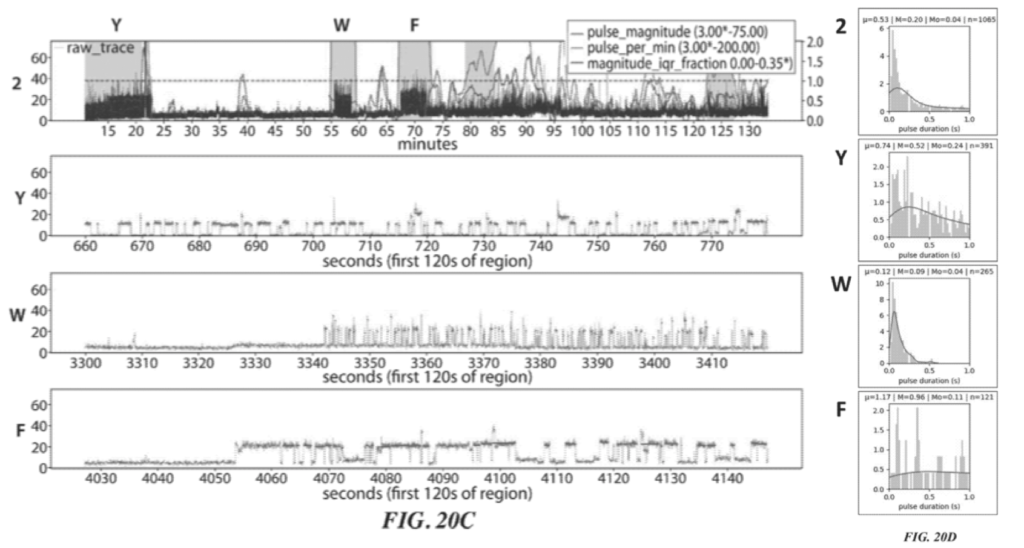
Example 6 shows what appears to be experimental data for this process. The example uses ATTO 542 label ClpS2. and an aminopeptidase (VPr). The protein they are attempting is sequence is YAAWAAFADDDWK.
ClipS2 binds to Y,W and F and apparently doesn’t bind to other terminal amino acids.
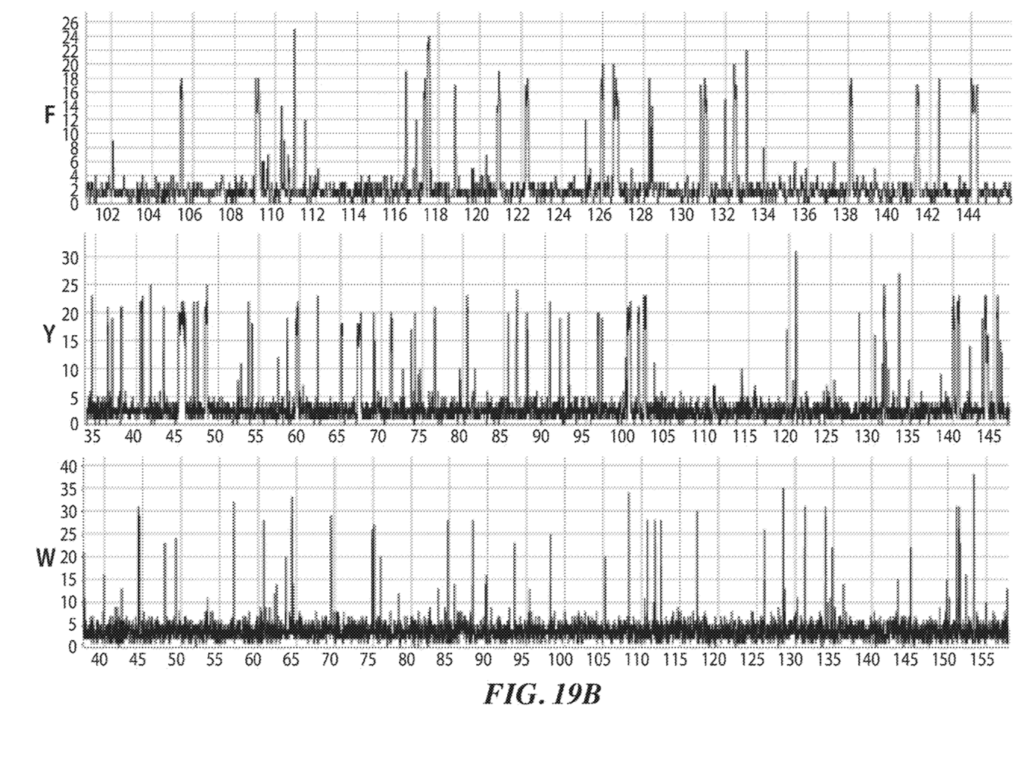
Two raw traces for this sequencing experiment are shown:
These two figures (20A and 20C) show two independent sequencing runs. Transient binding of ClpS2 to Y, W and F is shown. The experiment starts with the Y exposed as the terminal animo acid. As such we see transient binding of ClpS2 to Y from time point zero. At some point the cleavage enzyme comes in and the “Y” gets chopped off. “A” is now exposed at the terminal animo acid. ClpS2 shows no binding to “A” and we therefore don’t see any binding.
“A” then gets cleaved, exposing yet another “A” (still no binding). This is cleaved revealing W as the terminal base and we see transient binding again etc.
As it goes this is interesting, but it doesn’t really tell us anything about the sequence, which will naturally be runs of “F or W or Y” and not “F or W or Y”. It’s clear from the traces that the length of the transient binding period provides little informative information. For example, the time taken to cleave two “A” differs by a factor of 5 between the two experiments.
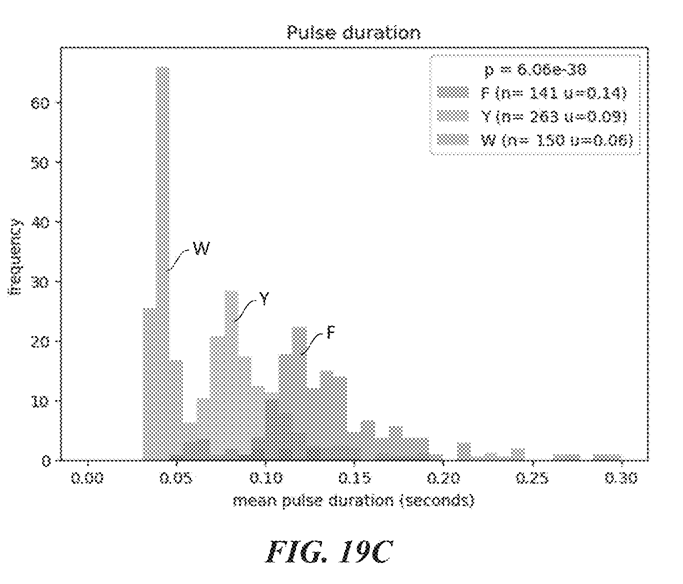
So in order to determine which of F, W, and Y was detected we need to look more closely at the transient binding. Figures 20B and 20D show histograms of the pulse duration. The variation and average durations seem to differ significantly for each animo acid.
This provides a basic proof of concept for animo acid detection. But it’s quite limited. The issues are as follows:
There’s no mechanism for detecting runs of identical amino acids.
The demonstration shows detection of 4 out of > 20 amino acids.
While the durations are somewhat distinct for these amino acids, it seems likely that 20 such distributions would show significant overlap.
The sequence is quite short (so we don’t know how long we can go without damaging the protein/other issues occurring).
Table 1 of the patent lists 33 amino acid recognition proteins, with 13 different binding patterns. These appear to cover 16 of the >20 amino acids ambiguously. A combination of there recognition proteins (and ideally a few more) might bring you closer to a full sequencing approach.
But, overall this work seems to provide a basic proof of concept of the detection process. With some additional work I could see this working as a protein fingerprinting technology. Where an ambiguous protein sequence is compared against a database of known protein sequences. In the above example the fingerprint might be something like:
“One or more Y”,”One or more not F,W,Y”,”One or more W”, “One or more not F,W,Y”, “One or more F”
With a long enough sequence, this may unambiguously identify a particular protein/class of proteins. However single point mutations might be more challenging.
Using additional recognition proteins would improve this fingerprinting process. Progressing this toward a sequencing platform seems challenging, and might require development of the technical approach.
Sequencing Approach 2 – Labeled Amino acid specific cleavage enzyme
Some data is also shown for a second approach. Here the cleavage enzyme is specific to certain animo acids. The demonstration shows that they have a method for incorporating labels into these exopeptides [5]. But there doesn’t seem to be any data demonstrating sequencing using this approach.
A list of amino acid specific exopeptides is provided [6]. However these seem to be far more limited than the recognition enzymes in approach 1. Only three types of specific exopeptides are listed those specific to Glu/Asp, Met or Proline.
This approach therefore seems more challenging and less developed.
Summary
Overall this seems like an interesting approach to a problem that hasn’t received that much attention. I’d expect the initial platform to be nearer to a “protein fingerprinting technology” than a full protein sequencing instrument. This seems like an interesting tool in its own right. If the initial instrument is framed as a protein sequencing platform, I would expect the error rate to be far higher than we’re used to seeing for DNA sequencing (probably in the order of >20%).
However, all this speculation is based on a single patent. They may have developed beyond this patent and it will be interesting to see what is finally released.
Notes
That’s it, you can stop reading now…
Ok, well… this section contains a few other notes from the patent. It doesn’t really add much to the discussion above, but they are here in part for my own reference. The footnotes below also support some of the assertions in the text above and may be of interest.
As is often the case, this patent mentions a number of other approaches which could be used. The patent discuss nanopore readout briefly. Indicating that the protein being sequenced could be immobilized on a nanopore. Recognition molecules (labels) are then detected through changes in conductance of the nanopore.
Other sections refer to “conductivity labels”.
Shielding elements. This is essentially a protein (or other element) that shields the recognition molecule from photo damage. Shield proteins are used in other single molecule sequencing approaches, and a number of methods are presented in the patent.
Other approaches to removing terminal amino acids are mentioned… Edman degradation, phenyl isothiocyanate.
Various other detection methods are briefly discussed for example Aptamers.
While I’ve not mentioned it in the text above there are some other nice plots of the pulse duration differences for ClpS2 in example 5:
Example 5: ClpS2 as recognition label. ClpS2 is labeled with dye. Single molecule intensity traces shown in figure 19B.
[1] “In some aspects, the application relates to the discovery of polypeptide sequencing techniques that allow both genomic and proteomic analyses to be performed using the same sequencing instrument.”
“Such strategies may require modification of an existing analytic instrument, such as a nucleic acid sequencing instrument, which may not be equipped with a flow cell or similar apparatus capable of reagent cycling. The inventors have recognized and appreciated that certain polypeptide sequencing techniques of the application do not require iterative reagent cycling, thereby permitting the use of existing instruments without significant modifications which might increase instrument size.”
[2] As always, a number of other approaches are also mentioned. But these are the approaches that have the best supporting data. I review some of the alternative approaches at the end of the post.
[3] One example shows proteins attached using a DNA linker, with 18% single protein occupancy. This seems lower than Poisson. Only single wells will be sequencable so this limits throughput. Example 2 (page 127).
[4] Amino acid recognition proteins. Table 1. Lists 33 amino acid recognition proteins and their preferred binding. Many of these appear to prefer the same amino acids. There are therefore 13 different types of “preferred binding” listed: FWY: 4, FWYL: 6, FWYLVI: 1, phosphorus-Y: 5, FWYLI: 1, KR: 1, DE: 1, KRH: 3, P: 2, KRHWFY: 5, PMV: 1, G: 2, A: 1.
[5] They reference the following paper, which uses non-natural amino acids. Chin J. W et al. J Am Chem Soc. 2002 Aug. 7 124(31):9026-9027
[6] Aminopeptidases. Table 3 lists amino peptidases. These should selectively cleave terminal amino acids. There seem to be 3 classes here with limited coverage of the amino acid space (Glu/Asp: 1, Met: 2, Proline: 6). Table 4 provides a much longer list of non-specific Amino-peptidases.
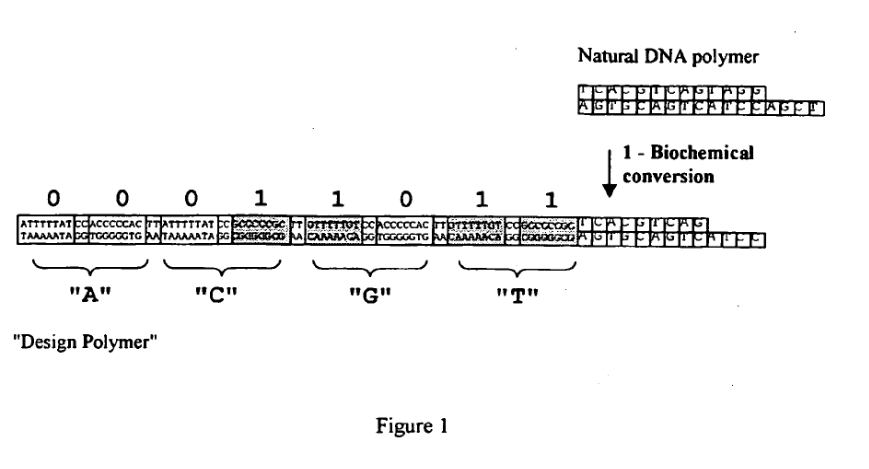
With the Stratos acqusition I wanted to write up a few notes on a much less well known (and now I think inactive) startup called LingVitae. LingVitae was a Norwegian single molecule sequencing startup. In many ways, their original “mission” was similar to Stratos’. They were working on a way to replace a single nucleotide with a magifying tag, or what is sometimes called a “Design DNA Polymer”.
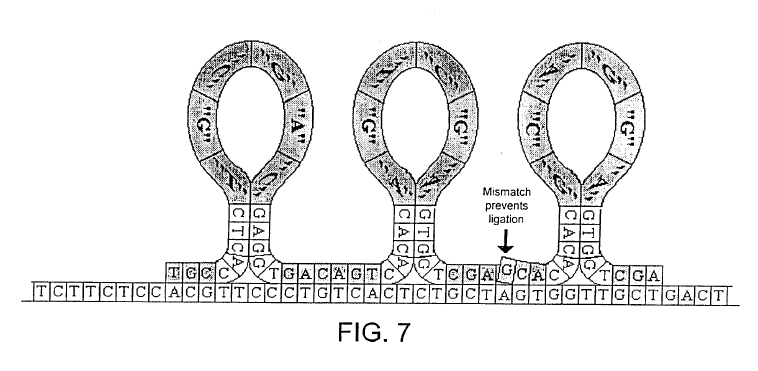
The approach to generating these “expanomer-like” strands, is rather similar one approach suggested by Stratos in their patents. Essentially loop/hairpin like oligos are hybridized to the template and ligated:
FIG. 7 shows adjacently aligned adapters which carry magnifying tags and which hybridize to the target and self-hybridize;
I’d guess there are a number of issues getting this to work. In particular, hybridization of such short (8mer) oligos might not be very specific. Will they really hybridize adjacently, specifically? Doesn’t the loop/bend cause a bunch of issues?
I couldn’t find anything that looked like experimental data in the patents. The closest I got was a 2007 paper describing the technique from Amit Miller [2]. This paper really just describes the concept and says “for proof-of-concept we designed and synthesized DNA oligonucleotides that encode”…”up to 8 bits of information”. Seems like they’re just synthesizing oligos, based on what LingVitae might have been able to produce. But there doesn’t seem to have been a followup paper showing any experimental work.
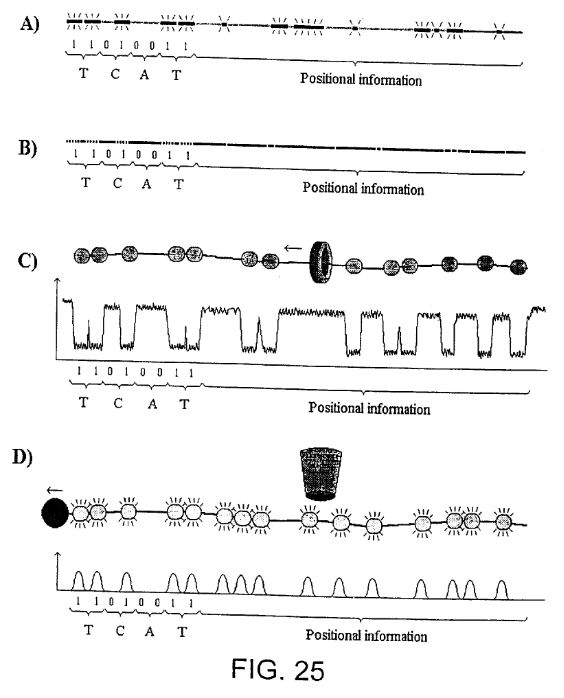
In their patents LingVitae, proposed a number of different read out methods, these include nanopore and optical approaches:
FIG. 25 shows examples of how signal chains may be used to obtain both sequence information (left) and positional information (right) in which A) shows a DIRVISH based method using fluorescence labelled probes that bind the target molecules in a characteristic pattern, B) shows an optical mapping based method in which the restriction pattern is used to give the position of the sequence, C) shows a method in which a characteristic pattern of DNA binding proteins are registered as they pass through a micro/nano-pore and D) shows a method using fluorescence labelled probes, proteins or the like which are registered as they pass a fluorescence detector.
LingVitae’s 2007 era website is shown below. This is from a time when they were patenting, and promoting work related to the design polymer idea. Their focus is on “high quality single molecule sequencing”.
This was before PacBio launched their instrument. So there were no “single molecule sequencing” platforms on the market at this time. LinkedIn shows about 30 former employees, so I imagine they had a reasonable team working on this.
Slowly LingVitae seem to have moved away from DNA sequencing. The website, and PR shifted toward the development of a cheap cellular imaging platform [1]. The idea was to use a DVD drive as a microscope. This was always part of the sequencing play, but sequencing gets downplayed from 2012 onward:
www.lingvitae.com started redirecting to discipher.co sometime in 2017? But it seems like the site itself was offline before that… then sometime in 2014 the website went offline completely. In 2017 it started redirecting to discipher.co:
Which as of ~May 2019 also appears to be offline. LinkedIn doesn’t show any active employees so I assume this is the end of the LingVitae story.
It’s a shame really, it would have been interesting to see preliminary data from the design polymer approach. If anyone knows what happened to LingVitae please get in touch!
From http://www.freepatentsonline.com/20070254280.pdf
Text from a version of their website, via the waybackmachine:
“Physical Magnification The units to discriminate in a biological DNA molecule are bases or base pairs with a size of 0,34 nm each. The units to discriminate in a Design Polymer are blocks of up to 25 bases or base pairs with a size of up to 10 nm each.
Maximalisation of Unit Differences The difference between the units to discriminate in a biological DNA molecule are only represented by a few atoms on a purine or pyrimidine skeleton attached to an identical backbone structure. The difference between the units to discriminate in a Design Polymer can be very significant and will be tailor made to achieve maximum resolution power on the read-out platform in question.
Binary Code There are 4 units to discriminate between in a biological DNA molecule and the read-out platform must thus be able to distinguish between 4 different levels or states. There are 2 units to discriminate between in a Design Polymer and the read-out platform must thus be able to distinguish between only 2 different levels or states or alternatively even easier use a very simple on-off approach
Removal of secondary structures A biological DNA molecule can take all forms of sequences and shapes and has a natural tendency to form secondary structures which can influence on the read-out process. A Design Polymer can be designed to avoid secondary structures and to ensure a reproducible behavior during the read-out process
Labels It is difficult to label every individual base in biological DNA molecule due to sterical hindrance. The repertoire of labels that can be used is thus limited. Incorporation errors, quenching of neighbor labels, etc. adds to the challenge. It is easy to label every individual unit in Design Polymer as the spacing between labels, unit sequence, and more can be designed. The repertoire of labels that can be used are thus almost endless. Incorporation errors, quenching of neighbor labels, and other challenges can easily be solved by smart design of unit sequences, spacing, and other Design Polymer parameters.”
“The whole purpose of the Design Polymer Concept is to enable read out technologies to perform rapid DNA analysis with superior quality and resolution, and after years of extensive research LingVitae is now developing its first series of Design Polymer products -DNA EXPLORER SYSTEM.
DNA EXPLORER SYSTEM is going to be a lab-on-a-disk based kit designed to convert biological DNA into synthetic Design Polymers. The intention is to provide read out companies with a powerful tool that enables them to obtain advanced “DNA molecules” designed solely for the purpose of single molecule sequencing.
The first generation of the kit will consist of a conversion jig and a set of four conversion disks. It is intended to be very user-friendly, so that anyone with basic lab skills should be able to perform the conversion of nucleic acid in less than 24 hours. “
“DNA EXPLORER Fluidics Station 500
Fluidics Station 500 will be the 1st generation of fluidics stations for processing the Conversion Disks.
The Fluidics Station 500 will incorporate advanced design that provides improved ease-of-use and true walk away freedom to dramatically improve efficiency in the end-users genetic analysis. The system will run unattended until completion of a Conversion Disk, freeing the operator to attend to other responsibilities, thereby helping to improve the workflow and operation of the laboratory.
Total processing time per Conversion Disk will be 6 hours and the Fluidics Station 500 is designed to operate in environments running 2-4 daily runs per system. A total of 4 runs will be needed for whole genome conversion of a eukaryotic genome. No dedicated or special power requirements.”
“DNA EXPLORER Conversion Kit
The Conversion Kit will contain a series of 4 Conversion Disks (Disk 1-4) for the Conversion of 100 Gigabases of 24mers from a target material as well as reagents needed for DNA purification and initial handling of the sample material.
The Conversion Kit reagents will formulate into the fewest number of individual components possible, reducing preparation and handling steps. All of the reagents will be ready-to-use solutions.”
Video linked from site: https://www.youtube.com/watch?v=zLb9ip2lOos
Picture from their site, describing the “lab on a dvd”:
My name is Nava Whiteford. I’ve worked for a few sequencing companies. I have equity in a few sequencing companies based on my previous employment (I try to be unbiased in my posts). You can contact me at: [email protected]