I’m curious to understand how cheap Illumina’s run cost (COGS) can get (as opposed to the cost per base). Illumina broadly have 4 classes of instrument. The iSeq, Miseq, NextSeq and NovaSeq. The NextSeq 550 seems like a Miseq++ and the NextSeq 1000/2000 a NovaSeq– (as the former lacks patterned flowcells, and the later doesn’t have the throughput of the NovaSeq).
The iSeq flowcells embed a CMOS image sensor. It’s likely difficult to get to a really low COGS here. The Novaseq uses patterned flowcells which they sell for >$10000 and likely have associated costs. So that leaves the Miseq.
The Miseq uses likely relatively inexpensive glass flowcell, and reagent cartridge. So I decided to take a look at that platform. The overall summary is that I’d guess at a lower bound a Miseq could be made for $5000, reagents likely cost at least $50.
Instrument
I purchased an old MiSeq camera from eBay. I can’t find my notes but I remember it used a Sony image sensor from a DSLR camera. Likely a monochrome version of the IMX038. This is built into a custom camera module with a Xilinx FPGA and associated memory. This is the same sensor used in the Nikon D90, which retailed for $900. I think it’s unlikely that Illumina could get this sensor the relatively low volume and integrate it with an FPGA and memory for less than this.
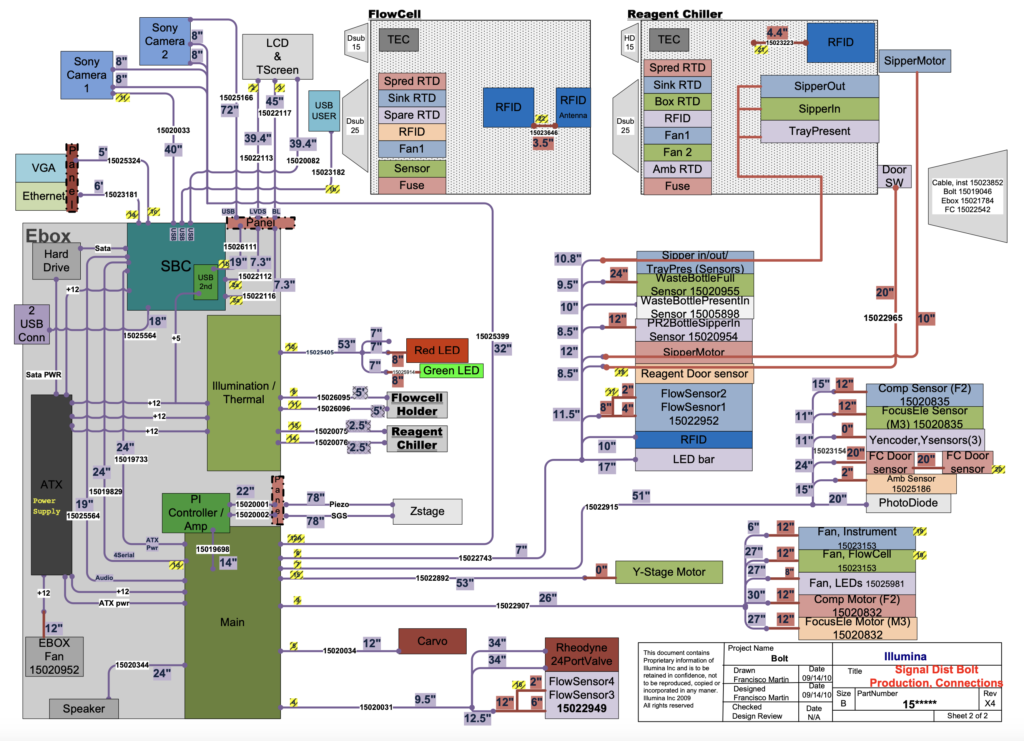
You can get a rough idea of what’s in the platform from this. There’s a PI Z stage. A Y-stage. Illumination uses LEDs rather than lasers. A bunch of TECs for temperature control. And some embedded compute (I’d guess x86/AMD64). Using this information my best guesses at the BOM cost for the most expensive components would be:
Cameras 2x Sony IMX038: Estimate $1800
Z-Stage (PI): $1000
Y-Stage: $100?
HDD: $50
Compute: $200
LEDs/photodiodes: $50
TECs: $50
Nikon x20 Objective: $400
Fluidics System: $300
Which gives us a total of $3900, for an instrument that costs the region of 50 to 100K (if you have exact pricing let me know). $3900 is a lower bound in my view, putting the instrument together in low volume likely adds significant overhead. Still I don’t see any reason why you couldn’t put together a MiSeq like instrument for around $5000 in high volume.
Flowcell and Reagent Cartridge
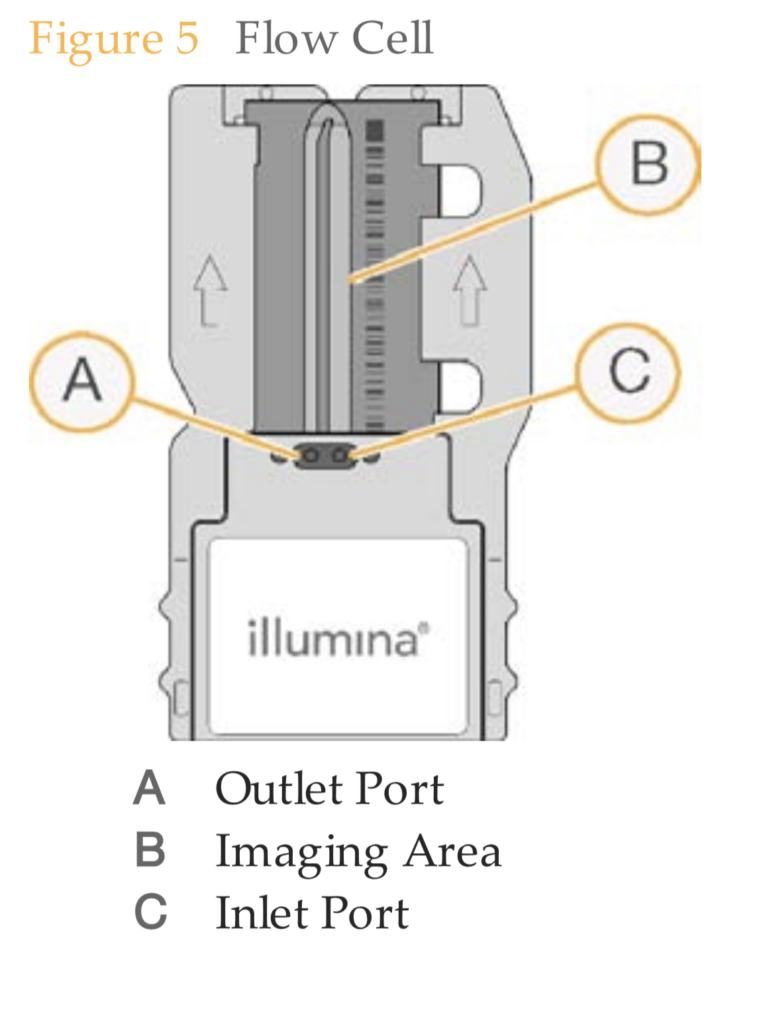
The Miseq flowcell seems to a simple glass substrate, with covalently bound oligonucleotides for capture. I’d be curious about accurate costings here, but I suspect it’s pretty cheap. Probably <$10. There are various companies you can buy flowcells from in relatively small quantities for not much more than this.
Which leaves us with the reagent cartridge:
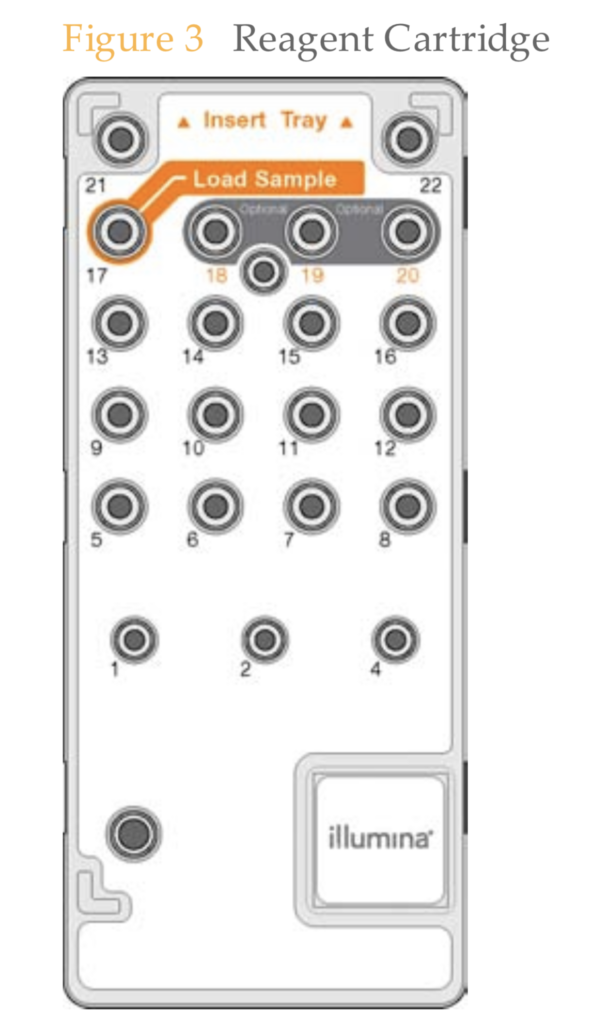
Digging through various bits of documentation it seems like we have 14 independent reagents in this cartridge:
1
IMT
Incorporation Mix
2
USM
Scan Mix
3
CMS
Cleavage Mix
4
AMS1
Amplification Mix, Read 1
5
AMS2
Amplification Mix, Read 2
6
LPM
Linearization Premix
7
LDR
Formamide
8
LMX1
Linearization Mix, Read 1
9
LMX2
Linearization Mix. Read 2
10
RMF
Resynthesis Mix
11
HP10
Primer Mix, Read 1
12
HP12
Index Primer Mix
13
HP11
Primer Mix, Read 2
14
PW1
Water
Most of these are either oligos (primers), polymerases, or nucleotides. None of which I imagine are particularly expensive. But it’s substantially more complex than the flowcell, and I imagine that QCing and putting this cartridge together along with the logistics around shipping it to customers is the dominating cost.
It’s hard to get accurate volume pricing on these reagents. But I suspect that it costs >$40. Which would put the cheapest MiSeq runs (~$500) within Illumina’s overall 90% margins on consumables.
Some was asking on Twitter about the effect of Illumina’s IP expiring on the sequencing industry. I figured I’d summarize my thoughts here. I’m not a patent lawyer, so you should consider that when evaluating my thoughts.
These two patents appear to be the fundamental IP required to build an “Illumina-style” DNA sequencer. Illumina have a number of polymerase patents. But my understanding is that the 9ºN (9 degrees North) and related polymerases incorporate modified nucleotides pretty well.
So the cluster and nucleotide IP are really the key to producing an “Illumina-style” DNA sequencer. As I understand it with that IP alone you could make something that looks very much like a genome analyzer 2.
I therefore would expect at the very least for “genome analyzer 2” style instruments to start appearing. The Singular platform looks like this to me, probably using alternate nucleotides until 2024… that is if they actually launch before 2024.
But they probably won’t be the only player to take this approach. At the very least MGI will be unblocked from selling instruments in the US. And I suspect companies like Element are working on something similar.
The question is, in what way does that fundamentally change the market. Well, for a start I expect it to put downward pressure on Illumina’s consumable pricing. At the moment there doesn’t seem to be any effective competition to Illumina. With these players appearing, with a similar data quality, and a similar error profile, I expect that many users will be able to switch platforms if they want.
Illumina makes ~10x on consumables. So… if these new players provide effective competition (which they should as the technology is basically the same) I expect there to be general downward pressure on consumable pricing, and we’ll see margins go down from 10x COGS.
This broadening of the market sucks for Illumina. And I suspect is why they’re shopping around for acquisitions. Pushing into other sequencing approaches (PacBio) and downstream into applications (GRAIL).
But how else will they respond? Well, I expect prices to do down, but I wouldn’t expect Illumina to solely compete on price. They’re iterated over the original IP pretty well and have a number of other advantages over new players specifically:
TDI Imaging
Two channel nucleotides/imaging
Patterned Flowcells
ExAmp
Super resolution
Better polymerases
This is all technology was developed for Hiseq’s and later instruments. It helps push throughout, reduce error rates, and increase read length.
So I expect Illumina to maintain a quality advantage. So one option they have is to push pricing as low as they can and have slightly better data quality (lets say, maxing out read length at 300bp as opposed to competitors being at ~100bp and having 90% >Q30 versus say 70%). Then everyone buys Illumina sequencers because they have the lowest cost and highest quality! Great! Except Illumina don’t make much money that way….
So I imagine what they’ll do is reduce pricing so that they’re competitive but not the cheapest option. Then they’ll let other players take the low end of the market and try and keep the high end where margins are higher (and users are willing to pay for slightly better data quality).
The above analysis relies on the fact that the COGS of competitors is probably going to be similar to Illumina. Of course there are other approaches on the market or in development. I’ve looked at most of these. Essentially for everything else available or on the horizon the data quality is not as good and COGS is higher than the Illumina approach. So I don’t expect some huge shake up here. As always there are niches to play in (base modifications, long reads etc.) and it’s possible those markets might grow.
But I think what’s really more interesting to me are approaches that reduce the COGS. In particular it’s been disappointing to see the limited impact of sequencing on SARS-CoV2 diagnostics. qPCR dominates here because the COGS for qPCR is essentially $1 and the COGS or instruments a few hundred. Even with multiplexing sequencing can’t yet approach this. So if you’d be interest in investing in (or codeveloping) such a platform, get in touch.
I’ve previously written about Singular Genomics. It now looks like their preparing for an IPO and have filed an S-1. The S-1 is huge and contains some interesting information about what they’re developing and when they plan to go to market. It’s fairly readable and I recommend taking a look if that kind of thing interests you.
In this post I’m going to try and quickly review the technological aspects of the Singular Genomics approach based on the S-1. This document doesn’t really go into specifics (and I’ll refer to other posts on this) but we can get a sense of where they lie technologically in respect to Illumina. So I’m going to dive into the technology and briefly review some of the business aspects at the end of the post. There are two aspects to the Singular Genomics play. The first is the basic sequencing instrument. The second are various sample prep and other analytical approaches they have in development, some of which complement sequencing.
Sequencing
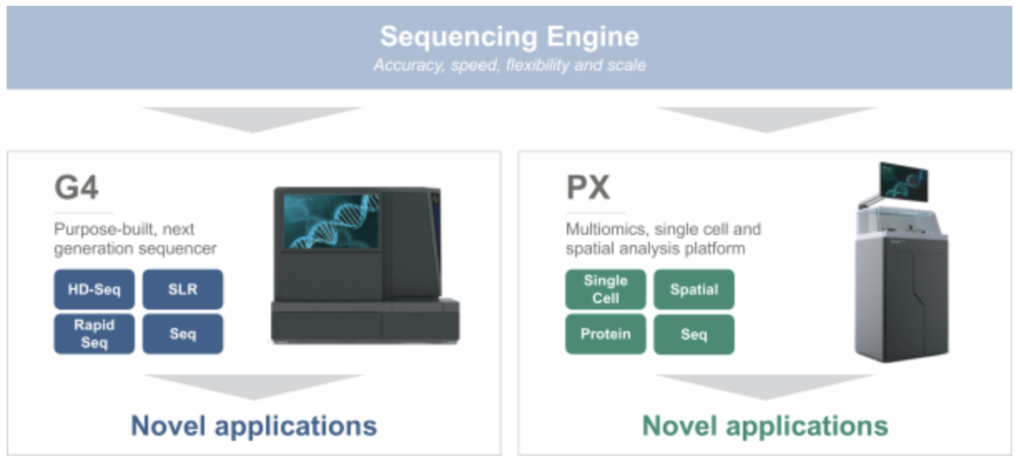
In summary, they look to be developing a 4 lane, (4 color?), Miseq-like instrument. There’s a figure buried in the middle of the document that describes the basic chemistry:
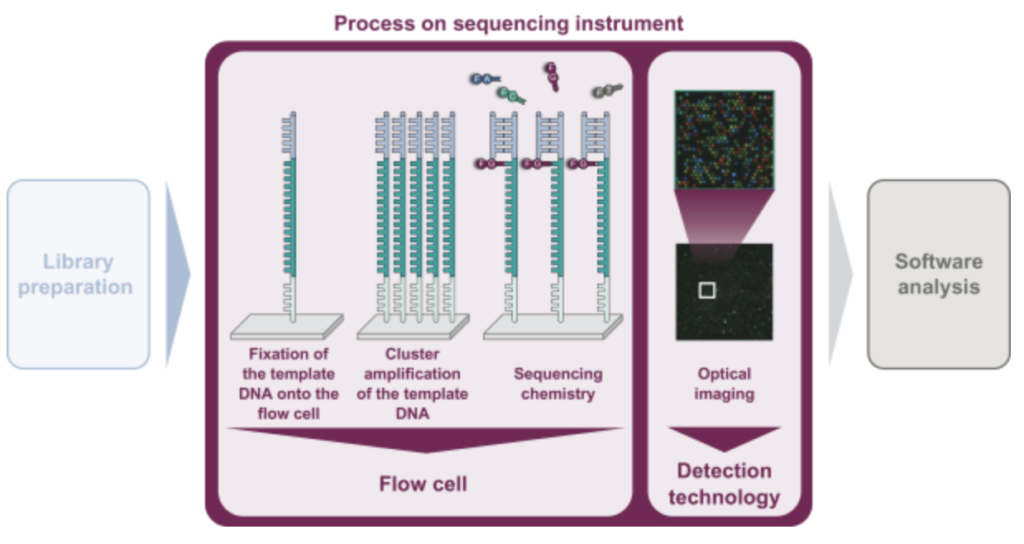
As in Illumina style sequencing, they perform on surface amplification of a single template to generate clusters, and then sequencing-by-synthesis to read out the sequence. The S-1 doesn’t state exactly how they are generating clusters. A quick patent search doesn’t yield anything either. Illumina use a bridge amplification approach, the initial IP was created by Manteia (acquired by Solexa, and then Illumina). I’ve discussed that IP elsewhere, and it appears to have expired. So my guess would be that they’re using this expired IP for cluster amplification.
They don’t appear to be using patterned flowcells, or ExAmp. This makes the approach more like that used on the Miseq (and original Genome Analyzer) than that used on the NovaSeq (and NextSeq 2000). This will limit the density of reads on the flowcell somewhat. From the image above it also looks like a 4 color chemistry, which complicates the optical system somewhat as compared to Illumina’s current generation instruments.
The S1 lends some weight to the idea that they are targeting “MiSeq-like” throughput: “We purposely designed our G4 Integrated Solution to target specific applications and to be capable of competing with other instruments across a range of throughput levels, particularly in the medium throughput segment.”. Flowcells, reagents cartridges, and instruments look fairly familiar:
The projected run times appear to be similar to a Miseq giving a “sequencing time of approximately 16 hours to complete a 2×150 base run.”. Similarly, Illumina quote a 17h run time for their Miseq nano kits at 2x150bp.
As I understand it the Illumina reversible terminator chemistry isn’t yet off patent. The S1 states that they “anticipate initiating an early access program followed by a commercial launch of the G4 Integrated Solution by the end of 2021, with intentions for units to ship in the first half of 2022”. Which suggests that they wont be using Illumina-style reversible terminators unless they believe that IP wont hold up anyway (this seems risky as Illumina are currently using it to block MGI from selling instruments in the US).
The S1 also states that “We in-licensed certain patents and other intellectual property rights from The Trustees of Columbia University” so it’s likely that they’re using Jingyue Ju’s nucleotides, as previously discussed.
They show some results on sequencing, overall data quality looks like it’s in the same ballpark as current Illumina systems. I’d suggest, much like MGI, it’s a reasonable drop in replacement for Illumina:
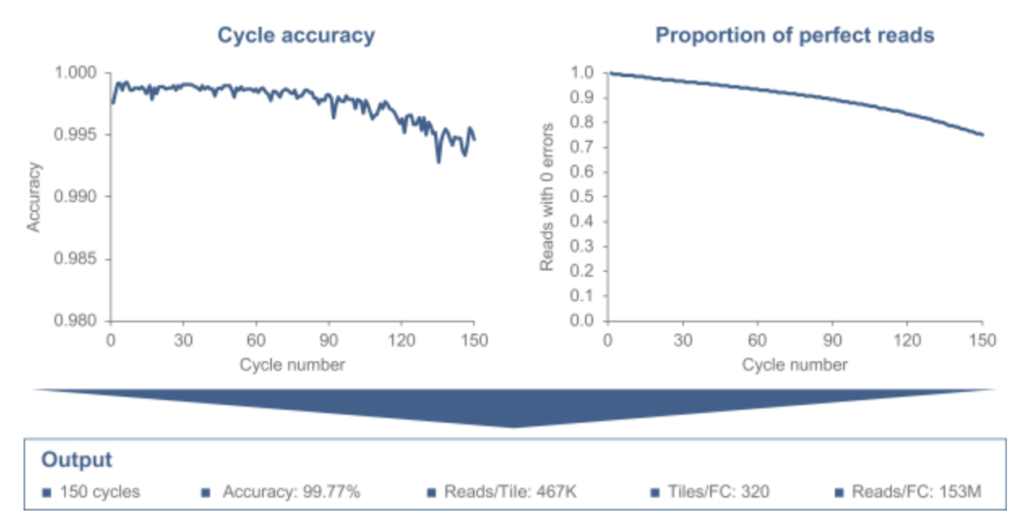
“This figure displays the current sequencing performance of our core Sequencing Engine with a demonstrated accuracy of 99.7% on 150 base reads (Q30 on greater than 70% of base calls) with a throughput of 153M reads per flow cell. We are targeting sequencing performance of Q30 on greater than 80% of base calls for 150 base reads and 330M reads per flow cell.“
Increased data quality though sample prep
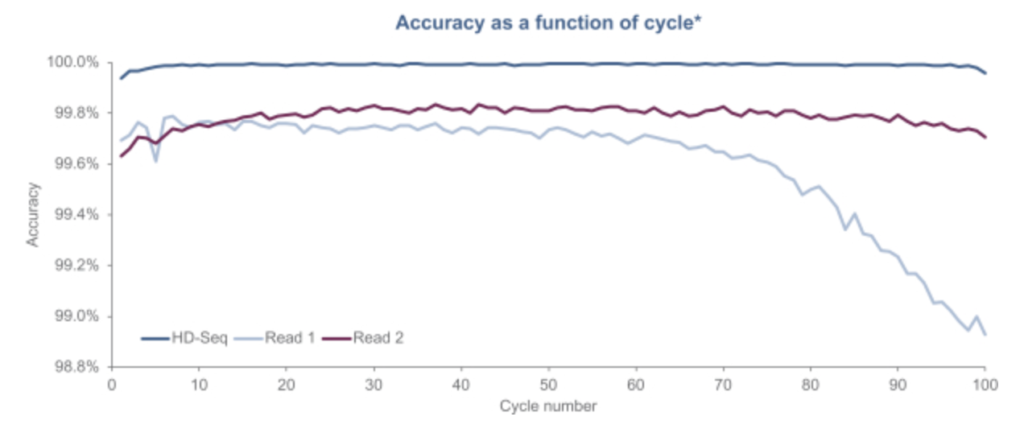
Another aspect of the Singular approach appears to be what they call “HD-Seq”. HD-Seq is designed to enable lower error rate reads that can “achieve accuracy levels of Q50” (error rates of 1 in 100,000). It appears to read out both strands of a double stranded fragment and combine the basecalls from both. They pitch this as being particular important for cancer diagnostics through the sequencing of cfDNA. And show some results:
They don’t state how this works exactly, but we can make some guesses. One way of doing this that’s been suggested is to tag the forward and reverse strand with the same index. You can then combine this information, giving you two observations of the same base, reducing the error rate. There’s a patent on this which I guess they may have licensed (though it’s not mentioned in the S1). Other approaches such as the 2D sequencing approaches used by Oxford Nanopore introduce a hairpin, allowing you to read through the forward, and then reverse strand.
You can also use a read pooling/indexing approach (such as 10X use to generate synthetic long reads) to make it easier to pair forward and reverse strands.
Beyond this, I could imagine a neat approach that takes advantage of cluster generation technology. Essentially, flow in double stranded DNA and weakly immobilize it on the flowcell. Then melt it such that the strands separate and attaches to nearby probes/primers on the flowcell. These two templates are then physically close on the flowcell. During image processing/basecalling you can then see that these two templates have a similar sequence (in reverse complement) and likely come from the same source double stranded DNA.
Singular state that their method can “provide higher accuracy than standard single-strand NGS sequencing methods (including ours)” so it’s likely agnostic of sequencing approach. Which suggests to me it’s likely an indexing or hairpin based technology.
Their motivation seems to be “oncology where there is an increasing need for higher sensitivity technology such as rare variant detection in liquid biopsy”. I’m not entirely convinced by this, beyond sensitivity, base modifications seem to be becoming increasingly important for early stage cancer detection. Is the difference between a single base accuracy of Q30 and Q50 critical? Or to put it another way, would you swap two Q30 reads for one Q50? Would be interesting to see more of a justification (or a reference) on the requirement for high quality reads.
Beyond this, they mask out potentially error’d bases: “the base call was only made if there was agreement in the base calls on the complementary strands”. So, while the overall error rate might be lower they limit their ability to detect individual SNPs… using this approach do you still retain the benefits of a lower overall error rate?
If a “read it twice” approach really is of critical importance to cancer diagnostics. There are a number of other techniques that would also work on Illumina’s platform (as discussed above). So this doesn’t feel like a key advantage of the Singular platform.
Other Stuff
Singular seem to be suggesting that they’ve developed a number of other analytical approaches and techniques and have a general purpose “multiomics” platform. The exact methods are not described. But they seem to be pushing further downstream into various applications. Many of which seem to be in 10Xs general territory:
Of particular note is their work on Single Cell and Spatial applications. They also mention Protein expression…
This is worth noting, but there not much in the S1 on the approaches used, and a quick search didn’t pull up any interesting patents here.
Business Stuff
That wraps it up for the technology. The S1 lists current investors, which contains many of the usual suspects:
Entities affiliated with Deerfield Private Design Fund IV L.P, Axon Ventures X, LLC, Entities affiliated with Section 32 Fund 2, LP, LC Healthcare Fund I, L.P, Revelation Alpine, LLC, Deerfield Private, Design Fund I.V., Domain Partners IX, LC Healthcare Fund I, ARCH Venture Fund IX, L.P., Axon Ventures X, LLC.
And that they’ve applied for the NASDAQ symbol “OMIC” which is a pretty cool symbol! They also state that they have 138 full-time employees (106 in R&D).
They state that: “Single cell, spatial analysis and proteomics markets: We are building our PX Integrated Solution to address the single cell and spatial analysis markets, which we estimate to be approximately $17 billion in 2021 based on available market data”. Given that 10Xs 2020 revenue was $298.8M. There’s probably something I don’t get here… where’s the rest of the single cell market?
——
So… those are my initial thoughts on the Singular S1. The basic sequencing approach looks very “Illumina-like” to me. There are a number of other plays like this around (e.g. MGI), and I suspect they will continue to put pressure on Illumina to reduce their consumable prices (which appear to be sold at ~10X cost of goods). But otherwise, I don’t see the approach as opening up new markets or giving us the ability to solve novel research problems.
DISCLAIMER: I own shares in various sequencing companies based on my previous employment. I consult in sequencing, and sequencing applications. And I’m working on a seed stage sequencing related project currently (get in touch if you might be interested in investing in such a project).
DISCLAIMER: I’m currently working on a seed stage sequencing related project. So, keep that in mind when evaluating my comments. And get in touch ([email protected]) if this might be of interest.
The press release doesn’t give much information, but references a recent publication, which provides more details. The paper describes a tiling array covering the entire SARS-CoV-2 genome. This is essentially a traditional microarray which covers the every possible SNP in the known SARS-CoV-2 genome. They describe this as follows:
“Here we describe a full genome tiling array with more than 240 000 features that provide 2x coverage of the entire SARS-CoV-2 genome and the use of such a genome tiling array to sequence the genome from eight clinical samples”
“Each base has two corresponding probe sets: one for the sense strand and one for the antisense strand.”…”one for each base”
So, Centrillion use a total of 8 probes per site. SARS-CoV-2 has an ~30Kb genome, which gives us the 240K features mentioned above. They use 25bp strands in their array. Tiling arrays have a number of limitations as compared to sequencing based variant detection. Firstly, it’s not clear what issues might be caused by multiple mutations covered by the 25mer. Such mutations would likely reduce hybridization efficiency, and could result in a variant miscall or “nocall”. The approach is also not able to detect deletions or insertions. These limitations make tiling arrays less interesting as a general purpose tool.
I personally would not characterize tiling arrays as “sequencing” or “resequencing”. In a tiling array you get a single read out for each position. Sequencing approaches generally produce a continuous readout of bases, without prior knowledge of the sequence context.
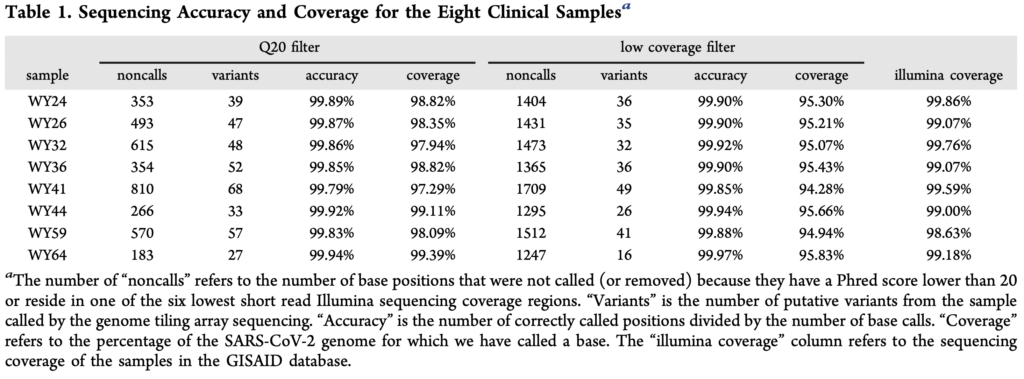
The paper presents a comparison of their approach against Illumina sequencing, summarized the in table below:
From the table above, noncalls are in the region of 1% seems to be one of the more concerning issues. I assume this means that both sets of probes essentially failed to provide a useful signal at these positions. The accuracy for the remaining positions seems reasonable (though less than I’d expect, given a known reference for the exact variant).
Summary
This is a fairly traditional tiling array for SARS-CoV-2 variant detection.
From the press release, it seems this is pitched as a lower cost alternative to sequencing. I don’t have good current pricing for microarrays, but I imagine the array they’re suggesting would cost >>$30, based on microarray prices I’ve seen. This is significantly more expensive than qPCR based testing (probably x10). This makes it too expensive for routine use in SARS-CoV-2 testing.
A detailed cost comparison against sequencing would be interesting. But I suspect we’re not talking about an order of magnitude cost difference. For me, this wouldn’t be enough to make an array based approach attractive, given that sequencing provides a richer dataset and more accurate.
Hopefully Centrillion will continue to develop some of their sequencing based ideas in the future.
My name is Nava Whiteford. I’ve worked for a few sequencing companies. I have equity in a few sequencing companies based on my previous employment (I try to be unbiased in my posts). You can contact me at: [email protected]