I was curious to know if there were any documented mutations which cover CDC Primers/Probes [1]. There’s work that has shown that mismatches in qPCR assays can “completely abolish PCR amplification” [2]. For diagnostic applications, mutations could mean that a qPCR based test would fail to detect SARS-CoV-2 or result in reduced sensitivity.
So, I downloaded all replacements (amino acid substitutions) from CoV-GLUE [3] [4]. I then extracted the nucleotide location identified in each replacement [5]. I then removed any duplicate locations. this resulted in a total of 3527 locations.
I then extracted the CDC primer sequences [6]. I wrote a small tool to do the following:
Load in the reference sequence, create a new sequence indicate mutation locations on the reference.
Find the location of the primer sequences on the reference [7].
For each primer, note where on the primer sequence there are mutations in the reference.
Report mutation location on primer.
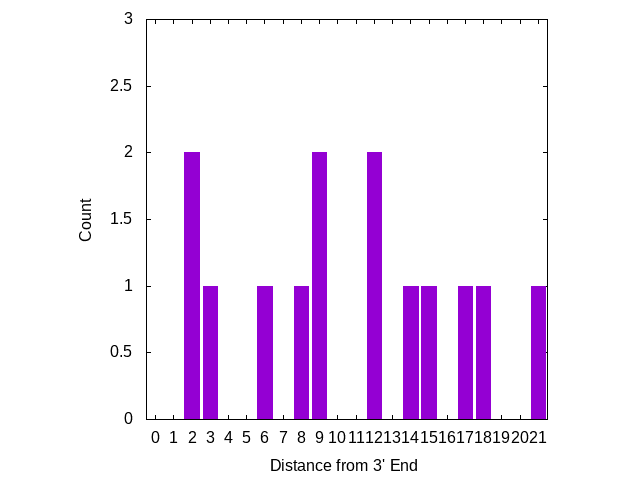
Mismatches near the 3′ end appear to be more significant, I’ve therefore plotted the number of mutations, based on there distance from the 3′ end of the primer. The plot below looks at primers only:
Three mutations are in locations which “may result in a 658-fold underestimation of initial copy number” [2] [8]. But there are no mutations on the 3′ terminal base, where a mismatch is likely to “abolish amplification”.
There does appear to be one mutation in the 3′ terminal base of one of the probes. However, I suspect terminal probe mutations are less significant than those in primers.
This analysis excludes far more common non-synonymous changes. I would expect these to be an order of magnitude higher. I would imagine this data is available somewhere, but I couldn’t see it in CoV-GLUE. Most likely it can be extracted from GISAID which seems to be the data source used for CoV-GLUE. If someone would like to work on an analysis of non-synonymous mutations, please get in touch.
Also, I’d warn again drawing any strong conclusions from the analysis presented here. This is very much a first look at the data and an attempt to feel out the issue. I think it would be interesting to replicate/build out this work however, and would love to hear any comments.
[4] Some messy scripts were required: http://41j.com/blog/2020/06/scripts-to-download-sars-cov-2-replacements/
[5] I used some awful awk to do this: for i in *; do awk ‘BEGIN{n=0;RS=”referenceNtCoord\”:\””;FS=”\”,\””;}{if(n==1) print $1;n++;}’ $i;done > mutlocs
[6] These are stored in the file called “primers”, in the tarball at the end of this post.
[7] This does a brute force alignment, looking for exact matches only on the forward and reverse strand. SARS-CoV-2 is only ~30Kb so computationally this is no problem.
Reading through some of Stratos’ more recent patents I came across a non-expandomer sequencing approach which I found quite interesting. The patent shows that Stratos had been thinking about other approaches to nanopore based sequencing. This suggests that the Roche acquisition may not just have been for their expandomer IP…
The Approach
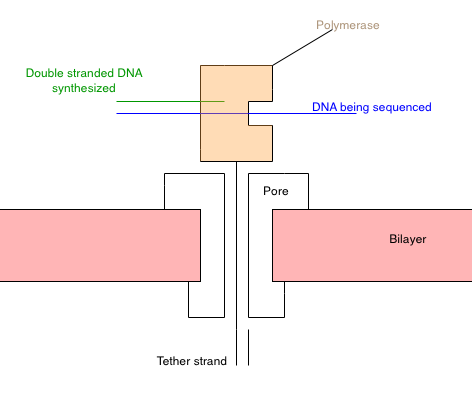
Schematic of my understanding of the approach presented in [1]. Also see their figure [6].
My understanding of the approach from [1] is summarized in the schematic above. Essentially you construct a polymerase [2] that has a “tether” attached to it. The tether is composed of a PEG repeat, which threads through a nanopore [3]. The PEG region has a short oligo on the end. Once it’s threaded through the pore, another oligo can be hybridized to it. This secures the tether in the pore.
With polymerase-tether complex in the pore, the system should look something like the diagram above. The polymerase is secured on the top of the pore. Due to its charge, I guess the polymerase would be pulled toward the pore, but it’s too big to pass through.
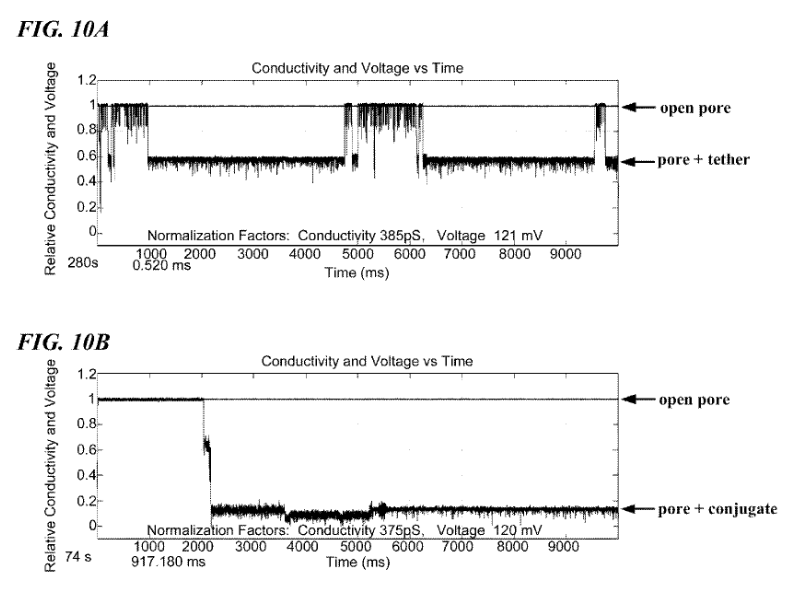
As in a standard Ionic/Protein nanopore setup, there’s a bias voltage, and ionic current passing through the pore. The polymerase is now blocking the pore. This causes a significant reduction in current flow. One of the figures in the patent illustrates this (and looks like experimental data):
FIG. 10A shows a signature electrical trace of an open nanopore and the nanopore partially occluded by a molecular tether. FIG. 10B shows a signature electrical trace of an open nanopore and the nanopore occluded by a DNA polymerase conjugated to a molecular tether.
Now, a template to be sequenced is introduced. Single stranded DNA enters the polymerase, and synthesis of the complementary strand occurs. However, the template DNA doesn’t interact with the pore directly.
The idea is that as the polymerase incorporates bases it will undergo confirmational changes. The patent suggests that these confirmational changes can be up to about 1nm.
The idea here is that different confirmational changes will cause the polymerase to block the pore with varying efficiency. So, for example when incorporating a base it might go through a series of confirmational changes, which result in less current flow, then more current flow, then back to baseline.
Ideally, each base would induce a distinct conformational change, and current blockage. The current trace can then be used to infer the sequence of the DNA template.
However, if you’re only able to detect incorporation/non-incorporation then nucleotides could be sequentially introduced.
The approach is somewhat reminiscent of that proposed by Roswell. Both of which seem to be trying to detect confirmational changes in the polymerase. In comparison to the Roswell approach, this seems like a simpler setup. The polymerase is also closer to the “sensor” which might make detecting confirmation changes easier.
Contrasting this with other ionic nanopore approaches, it’s kind of nice that the strand doesn’t go through the pore. Ideally in the Stratos approach there would be at most 4 different signal types, one for each base. Rather than the signal being some combination of all the nucleotides currently in the pore. Theoretically this could result in lower error rates.
Overall the idea seems at least plausible, and I’ll be curious to see how it plays out.
[2] A number of enzymes that process DNA could work (for example an exonuclease). And the patent states this, but a polymerase seems the most likely option.
[3] They talk about and show various protein nanopores. But I imagine a solid state nanopore may also be viable, particularly as the dimensions of the pore maybe slightly less critical in this approach.
[4] “The tethers were constructed of three domains (i.e., “segments”): 1) a polyethylene glycol (PEG) repeat region, located proximal to the polymerase and designed to span the nanopore channel; 2) a short oligonucleotide, designed to hybridize to a single-stranded oligonucleotide on the opposite side of the nanopore relative to the polymerase to anchor the assembly; and 3) a negatively charged phosphoramidite tail, located most distal to the polymerase and designed to facilitate threading of the tether through the nanopore. FIG. 9 is a SDS/PAGE gel that shows the size of the unmodified KF polymerase (lane 1), the KF-tether 1 conjugate (lane 2) and the KF-tether 2 conjugate (lane 3). As expected, the conjugates show an increase in mass compared to the unmodified polymerase.”
[5] “When this polymerase complexes with a nucleotide that is the complement to the template base in the next extension position the polymerase reconfigures into what is referred to in the art as a “closed” conformation. At a more detailed structural level, the transition from the open to closed conformation is characterized by relative movement within the polymerase resulting in the “thumb” domain and “fingers” domain being closer to each other. In the open conformation the thumb domain is further from the fingers domain, akin to the opening and closing of the palm of a hand. In various polymerases, the distance between the tip of the finger and the thumb can change up to 10 angstroms between the “open” and “closed” conformations. The distance between the tip of the finger and the rest of the protein domains can also change up to 10 Angstroms. It will be understood that this change will be exploited in a method set forth herein.”
[6] This figure in the patent is supposed to show the threading/polymerase. But I find it a bit confusing.
Other quotes….
“The present disclosure relates to methods and constructs for single molecule electronic sequencing of template nucleic acids. The constructs are molecular sensor complexes which comprise a processive nucleic acid processing enzyme localized to a nanopore. Conformational changes in the enzyme induced by single nucleic acid processing events are transduced into electric signals by the nanopore, which are used to identify individual nucleotides. The methods can include the steps of providing a membrane with the nanopore and the enzyme complexed with a template nucleic acid localized proximal to an opening in the pore, contacting the enzyme with an ion conductive reaction mixture including the reagents required for nucleic acid processing, providing a voltage drop across the pore that induces ion current through the pore that is modulated by conformational changes in the enzyme, measuring current through the pore over time to detect nucleotide-dependent conformational changes in the enzyme, and identifying the type of nucleotide processed by the enzyme using current modulation characteristics, thus determining sequencing information about the nucleic acid molecule.”
“The polymerase is secured to the pore by hybridizing a short oligonucleotide anchor to the tether construct on the distal side of the nanopore.”
“These results indicate that a tether and a tether-polymerase conjugate can be anchored to a nanopore and, moreover, that the resulting complex can generate reproducible electrical signals. Polymerase-nanopore complexes are thus capable of modulating current flow through the pore and show promise as useful sensors to transduct mechanical events into electrical signals.”
“One or more of the transitions that a polymerase undergoes when adding a nucleotide to a nucleic acid can be detected using a molecular sensor complex as described herein.”
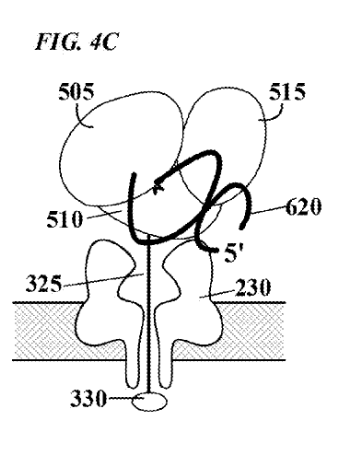
“FIG. 4C depicts the polymerase in a second, ™, “closed” configuration, which is induced, e.g., by binding of incoming nucleotide 605 to form a correct base pair with the template nucleic acid. In this second configuration, the degree to which the enzyme physically occludes the pore is reduced, and consequently the flow of current through the pore will increase. Such modulation of current flow generates an electronic signal specific for nucleotide species”
“In one embodiment, each of the four nucleotides induces a different polymerase conformation, as illustrated in FIG. 4C. . The movement of the polymerase during the incorporation of a nucleotide will modulate the ion current through the pore in a characteristic and reproducible manner, generating a signature electric signal.”
“In another embodiment, the average amplitude of the current modulation doesn’t change, but rather the noise in the current modulation changes as a single nucleotide is bound and incorporated. In yet another embodiment, the current modulation system only indicates an incorporation event but does not discriminate the base type. In this embodiment, the sequence information about a nucleic acid is obtained by sequentially flooding the senor complex with one of four reaction mixtures containing one of the four nucleotides and detecting the presence or absence of an electric signal.”
“For proper function of the molecular sensor complexes of the present invention, it is necessary that the enzyme be stably localized to the pore in sufficiently close proximity to reliably influence, or modulate, current flow through the pore. Several alternative localization and/or attachment structures or compositions are contemplated by the present invention, some which are illustrated schematically in FIGS. 5-8. FIG. 5A depicts one embodiment in which enzyme 500 is localized to pore 220 by covalent attachment to tethering structure 325, herein referred to simply as a “tether”. Tethers may be designed to thread through the lumen of the pore, from one side of membrane 100 to the other. Tethers may comprise one or more structural domains, or “segments”, designed to perform one or more functions.”
I’ve previously written about Apton Biosystems. When I wrote that post there wasn’t much to go on. However, a patent [2] has recently been published which reveals a bit more information.
The motivation stated in the patent is that “to reach a $10 30× genome”…”the amount of data per unit area needs to increase by 100 fold”. Elsewhere in the patent they mention that the prior art is a pitch of 1 micron. HiSeq wells were ~500nm. So they want decrease well size to ~100nm.
This premise, is slightly shaky as Illumina flowcells and reagents are sold at significant profit. I imagine a large part of Illumina’s costs are related to logistical issues, rather than consumables themselves.
In any case, the patent proposes a massive cost reduction by more densely packing DNA on the flowcell. The patent mostly refers to ordered arrays, and many examples refer to a single molecule approach. The basic chemistry however seems to be pretty standard Illumina style sequencing-by-synthesis.
The figure below shows a simulation of DNA attached to a surface, at varying pitch (spacing). The right-hand images are de-convoluted versions of the left. It’s clear that as the pitch gets smaller, the image gets more crowded, and it’s harder to identify individual spots.
When imaging using a standard optical microscope, you would expect your density to be diffraction limited. Essentially, you can’t clearly identify features smaller than the wavelength of light (~200nm)… normally.
However, a number of recent techniques have broken the diffraction limit. These have allowed optical microscopes to resolve features down to 10s of nanometers. In this patent, Apton apply some “super-resolution”-like approaches… but in a limited scope (we’ll revisit what Illumina might be doing here later).
A basic super-resolution approach is shown below (not from Apton):
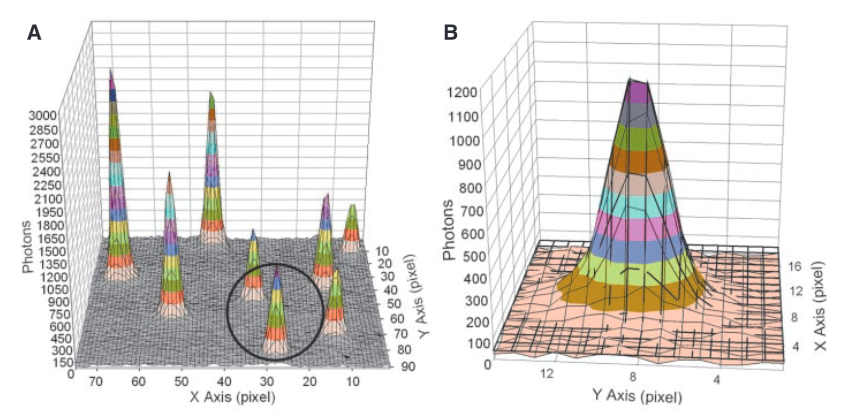
From [1]. The images above show the signal detected from a individual fluorophores. Each pixel is 13um, using 150x magnification this covers ~86nm on the surface. To generate super-resolved locations they do a Gaussian fit/find the fit of the intensity registered from a single fluorophore.
Each “peak” in part A of the figure above is the signal from a single fluorophore. Because the peaks are well separated we can extract each one and look at its distribution. In part B we see a single distribution. This is a 2D Gaussian. If we just took the pixel of highest intensity as the location of the fluorophore our resolution would be diffraction limited to ~200nm. However, by performing a Gaussian fit over the distribution we can determine the location to sub-pixel resolution. In this case, they could identify fluorophore locations at a final resolution of 1.5nm.
The above approach only works because the flurophores are well separated. If the Gaussians overlapped, the fit wouldn’t work. In the image above you can see the FWHM of the Gaussian is about 3 pixels, this represents ~250nm on the surface. I’d imagine if flurophores were any closer than this you’d have issues.
In their patent, Apton use the above approach to identify positions of single DNA strands on the surface to a sub-diffraction limited resolution of “10 nm RMS or less”. Apton appear to use essentially the above approach. However they have a problem, they want to pack the molecules as closely as they can to improve density. This means they are not well separated like those in the figure above.
To get round this Apton seem to use a couple of approaches. The first is that they use signals from multiple cycles to identify molecule positions. If the oligos attached to the surface have a fairly random distribution of bases (like the human genome) this should help a lot. For each molecule, you can select a cycle where it is illuminated, but none of its neighbors are. This means there is no crosstalk at this position and you should be able to get a good estimate of its position.
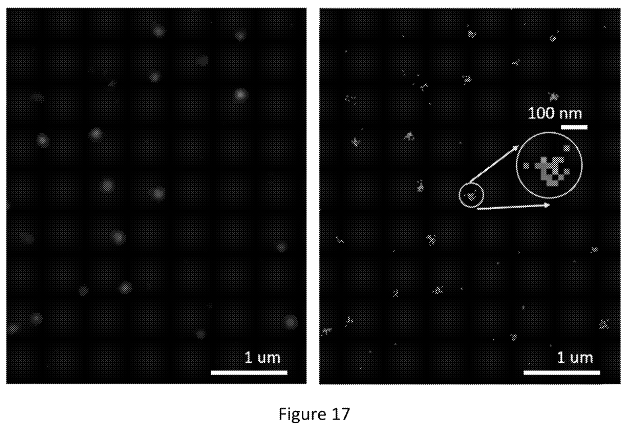
The figure below shows what appear to be images from a real single molecule experiment:
“right panel, shows each peak from each cycle overlaid. The left panel is the smoothed version of the right panel. Each bright spot represents a molecule. The molecule locations are resolvable with molecule-to-molecule distances under 200 nm.”
The right image shows positions identified from different cycles piled up (super-resolved positions I assume). In the left image they’ve used these to create another Gaussian. I would guess they then take the peak of this second Gaussian to give the final location of the molecule. This way, they can incorporate information from multiple cycles to give themselves the best estimate of the molecules location.
Using the above process I’d imagine they can get pretty good spot locations. They also mention the use of a crosstalk correction algorithm during the location identification step. But I’d imagine just filtering out bad looking Gaussian would work reasonably well.
While they may now have good spot/molecule locations this doesn’t mean they can pack DNA at ~10nm on the surface. This is because in any given cycle, there will be adjacent molecules which are fluorescing. The resulting Gaussian PSFs as imaged will overlap meaning that spots can’t be resolved. This is essentially crosstalk between adjacent spots.
Apton appear to be trying to use there super-accurate spot locations as the input to their crosstalk correction algorithm. The crosstalk correction process isn’t described in detail. But I can see that with very accurate spot locations, you can parameterize a model to which you can fit your observed signal.
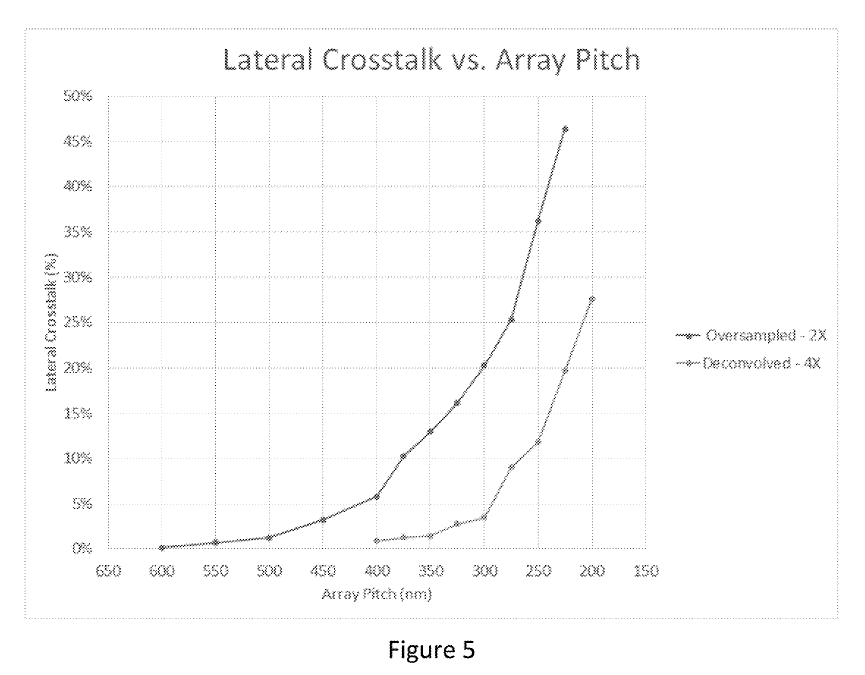
This sounds great, but crosstalk seems to increase exponentially:
The examples say “molecule locations are resolvable with molecule-to-molecule distances under 200 nm” and elsewhere they say “acceptable crosstalk levels” … “occurs for pitches at or above 210 nm”.
So it seems based on this, a pitch of ~200nm is viable, but it’s not clear that you can go lower than this. This seems unfortunate, as it’s only about a quarter of the size of Illumina’s wells.
What About Illumina?
As mentioned above, super-resolution has been around for a while. In fact, the Genome analyzer 2 used super-resolution-like techniques. Cluster locations could be identified to sub-pixel resolution. Rather than just picking the “brightest” pixel in a cluster, adjacent pixel intensities could be fitted to a PSF to give a more accurate cluster location.
Illumina appear to have now filed a bunch of patents on various approaches to increasing density. One patent uses a DNA-PAINT [3] approach, which they suggest can increase the packing density to “may be less than about 20 nm”. Another describes a STED approach [4] (200nm).
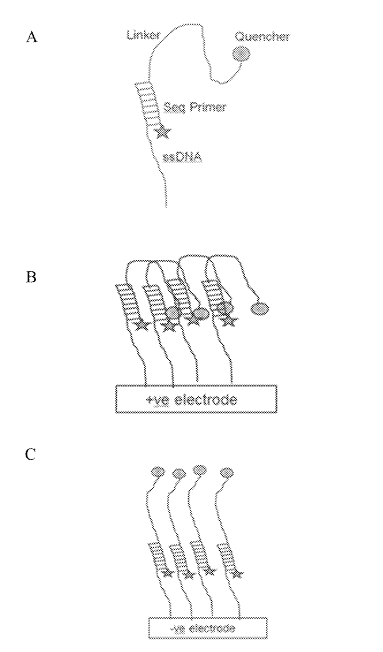
There was however one patent that I found quite fascinating. This approach appears to be for an iSeq-like [6] platform where clusters sit over a CMOS image sensor. The device incorporates additional electrodes which allow an electric field to be created under the cluster.
This field can then be use to electrically deactivate a fluorophore (in one example, by pulling a quencher down on to it):
This seems very neat. Essentially it would be that you could image one cluster while quenching all its neighbors. This would remove any crosstalk. Giving you good separation of signals while retaining density. Theoretically you could push clusters very close together.
While the Illumina patents describes some interesting approaches, I didn’t see anything that looked like a real experimental setup or real datasets. So, maybe much of this is theory at the moment. I guess we’ll have to wait an see!
References and Notes
[1] Myosin V Walks Hand-Over-Hand:Single Fluorophore Imaging with1.5-nm Localization.
[6] I don’t see any reason why a similar setup might not be used with a normal (patterned or otherwise) flowcell with embedded electrodes. But the patent seems to focus on a iSeq-like apporach.
I wanted to download a set of mutations in SARS-CoV-2. CoV-GLUE seems to be a reasonable database of mutations in SARS-CoV-2. However the web interface doesn’t seem to have an option to download a dataset. And there isn’t a published API. So I threw together some ugly bash/awk to get what I wanted. I don’t imagine this will work for long, as the website appears to be under active development. But here are my notes anyway.
The website works off a (undocumented?) JSON API. I used the follow JSON template to get replacements (non-synonymous substitutions) which occur in 2 or more sequences:
{"multi-render":{"tableName":"cov_replacement","allObjects":false,"whereClause":"(true) and (((num_seqs >= 2)))","rendererModuleName":"covListReplacementsRenderer","pageSize":500,"fetchLimit":500,"fetchOffset":FETCHOFFSET,"sortProperties":"-num_seqs,+variation.featureLoc.feature.name,+codon_label_int,+replacement_aa"}}
The above goes in a file called templ. I then just modify “FETCHOFFSET” using sed and download the first 4500 mutations (at the time of writing there are 4000 odd mutations. You’d want to stick this all in a loop… but I didn’t bother:
The CoV-GLUE database seems like a great resource. I hope they add a feature to download sequences/results soon. I’ve seen database results presented in a few preprints. It would be nice it those papers could also include the raw data, otherwise they’re unfortunately going to end up being difficult to replicate…
My name is Nava Whiteford. I’ve worked for a few sequencing companies. I have equity in a few sequencing companies based on my previous employment (I try to be unbiased in my posts). You can contact me at: [email protected]