This post previously appeared on my substack.
I was recently asked about Nooma Bio. This is a company and approach I’ve not looked at for some time. But it appears they published a couple of papers at the end of 2019. So here I’ll review the company and their 2019 publication.
I recently wrote about Solexa and much like that company, Nooma’s journey has taken a somewhat meandering coarse. The company was originally founded as TwoPoreGuys by Daniel Heller and William Dunbar in 2011. Dan came from a background in tech, having previously founded email software company Z-code which was acquired for ~9.4MUSD in cash and stock back in 1994.
Dan left TwoPoreGuys in 2018, and in 2019 the company turned into Ontera. Murielle Thinard-McLane took over as CEO. Nooma.bio was then founded in 2020, as a spinout of Ontera, and retains the same CEO and CTO.
Both companies are pursuing ionic solid state nanopores detection platforms. But from what I can gather, it’s Nooma that’s taking forward the original two pore approach. And this is what I’ll be discussing here.
Two Pores are better than One?
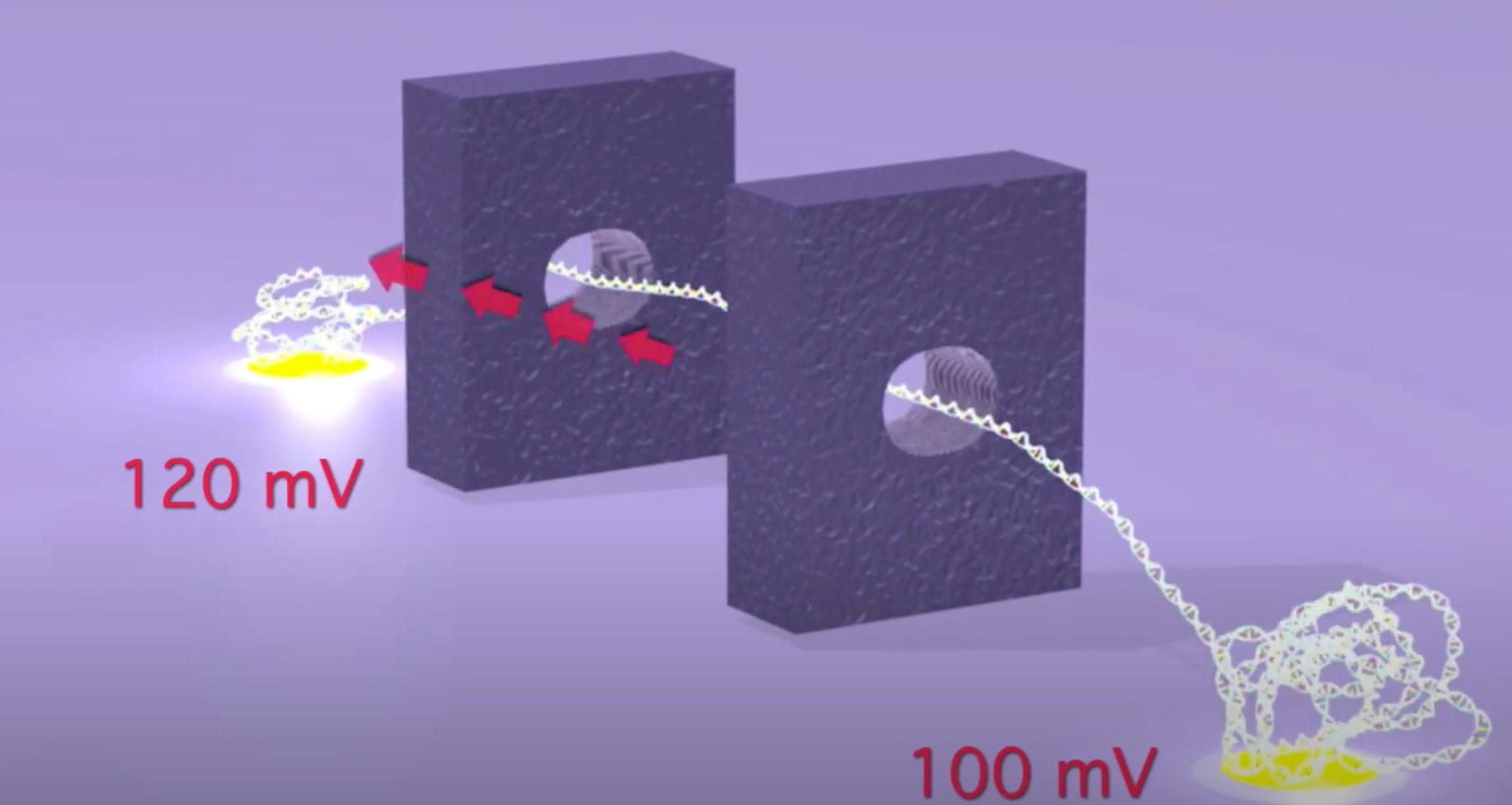
As originally presented the TwoPoreGuys’ approach used two nanopores and a small difference between two larger bias voltages across these pores. The original website, is unfortunately long gone, but the youtube videos are still up:
As presented, this never made much sense to me. The above seems electrically the same as setting a 20mV bias voltage. The two pores in this original approach were also purely for motion control, the idea was that there would be other sensors (they give the example of tunneling current electrodes) in the gap between the pores.
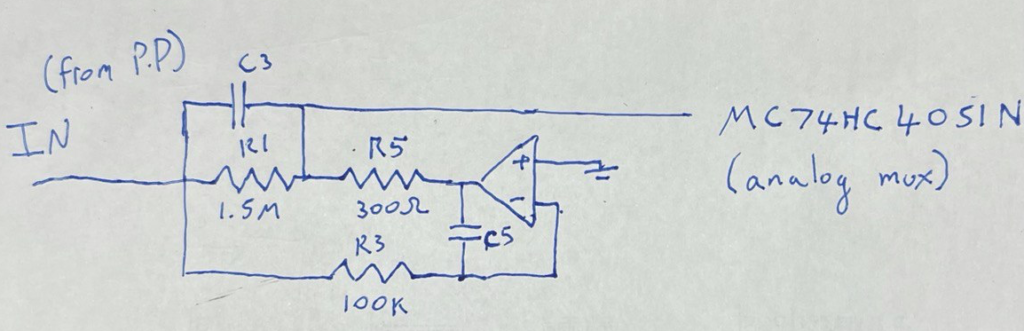
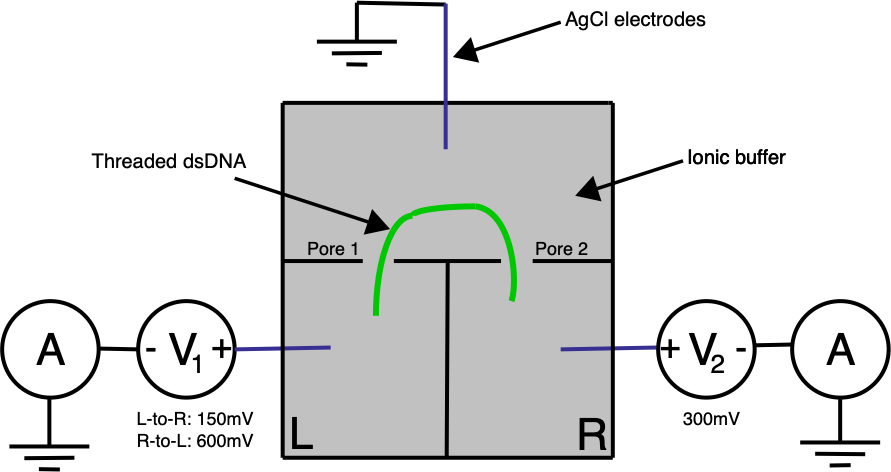
Ontera’s (pre-Nooma) 2019 paper takes basic approach in a slightly different direction. Here they use two adjacent pores on a planar substrate, in a three electrode system:
A double stranded (negatively charged) DNA translocates toward the most positive electrode. By changing the bias voltage at V₁ we can the most positive point either V₁ or V₂. All our ionic currents on the other hand will flow between V₁ / V₂ and our ground point which is in the chamber between the two pores.
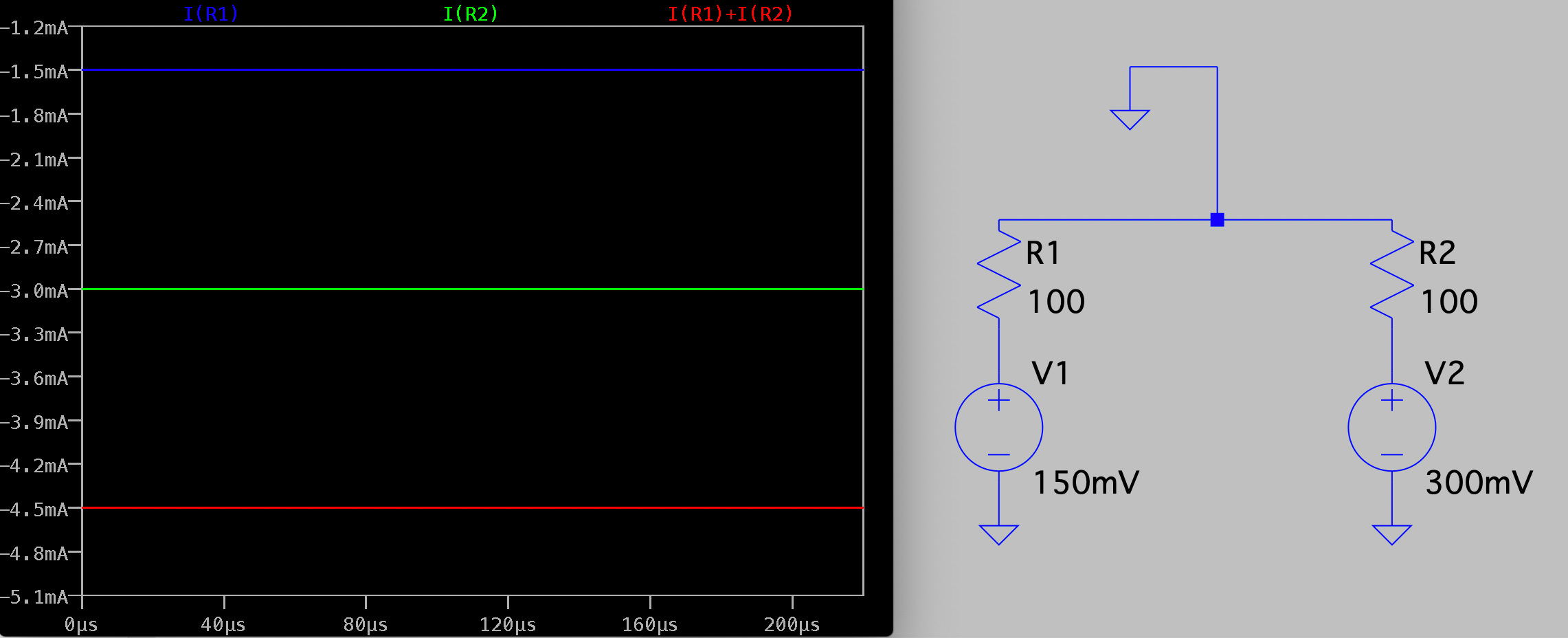
Electrically the system is pretty simple. In the diagram below R1 and R2 represent our two pores, and in reality are in the Teraohm range:
This approach gives us three principal advantages over a single pore system:
- We can sense the DNA twice (once as it passes through each pore).
- We can flip the voltages around to reverse the DNA translocation.
- We might get better motion control by confining the DNA between two pores.
In particular the ability to “floss” a single strand backward and forward through the pores gives multiple observations of the same molecule. You can of course also do this with single pore systems, but the author’s suggest that the two pore approach helps maintain the strands orientation.
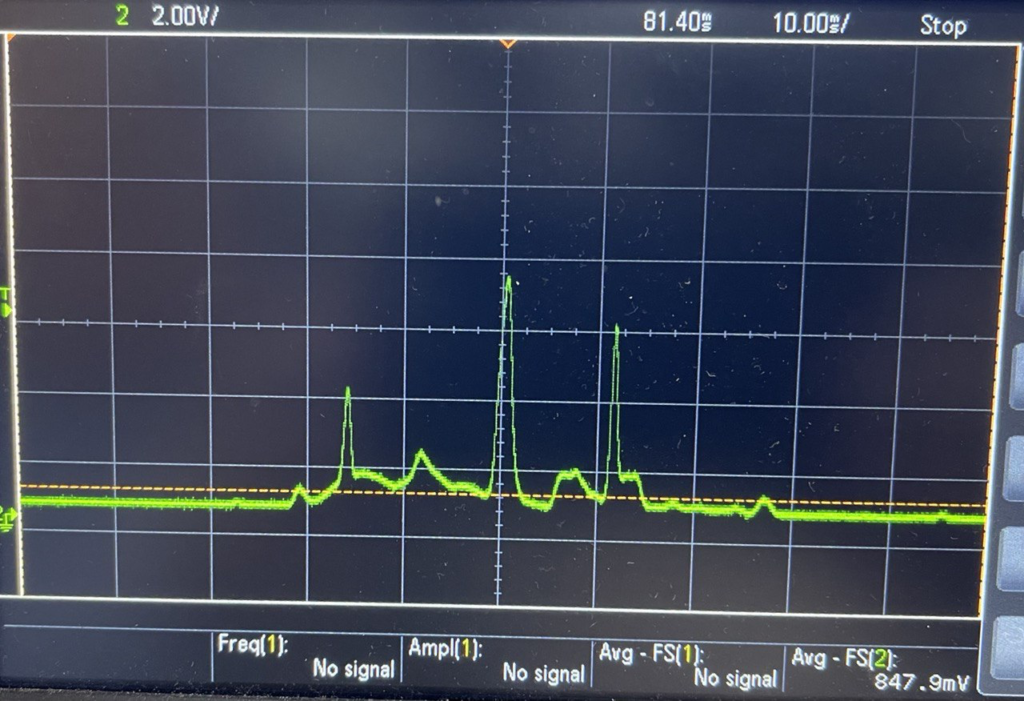
To demonstrate this in their 2019 paper they bind a couple of Streptavidin protein tags to Lambda DNA. As these tags pass through the pore they show sharp dips in current as they block the pore. As soon as two tags have been detected, the voltage is flipped and the DNA will translocate in the back through the pores, in the reverse direction.
They can floss the same strand hundreds of times. However, it seems like there’s a fixed probability a tag will be miss registered, and the strand being ejected from the pore. This results in an exponential distribution of events/per strand (37% of the events had less than 5 scans):
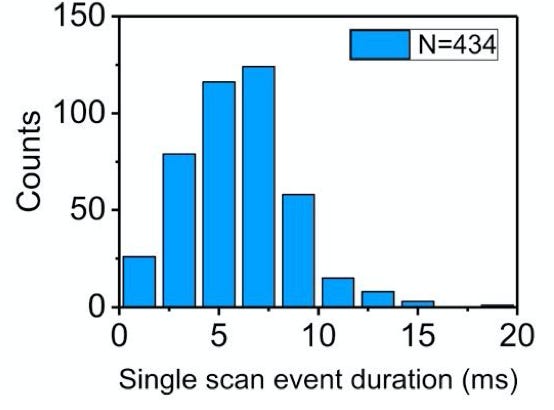
While the individual scan duration (which I think is a rough proxy for tag-to-tag dwell time), looks roughly poisson:
The paper shows a number of different experiments, with up to 7 tags at nick sites on Lambda DNA.
Thoughts
The approach described might be usable for mapping applications. By averaging the multiple “flossed” observations you can get better estimates of the tag to tag distance. The resolution here seems like it’s on the order of a few hundred nucleotides.
So, you can imagine a platform where you nick and tag DNA and read it on a 2 pore platform. The problem is, that we already have pretty reasonable mapping tools. And the market (~$10M?) may not justify the development costs.
Unfortunatley, to me the approach doesn’t seem to be compatible with DNA. The method doesn’t slow translocation sufficiently to be able to detect single bases. In the paper, they use a 10KHz bandwidth, and Lambda strands seem to translocate in ~10ms. Which is less than 1 data point per base.
And if DNA is problematic, using this as a nanopore protein sequencing platform is likely even more challenging.
Increasing the bandwidth much beyond 10KHz isn’t very practical, and it’s not clear that you can slow the translocation (particularly of single stranded DNA) much more using this approach.
In any case, solid state pores have not yet reached feature sizes where DNA sequencing becomes practical, and this 2pore approach doesn’t seem like it would be compatible with protein nanopores and enzymatic motion control.
I do like the fact that you’re precisely stretch the strand between two points, and that you can obtain information on the strand from a first pore, before it translocates through a second. There’s one patent from Nooma discussing Material Sorting Applications which seems like an interesting idea, that could take advantage of this unique feature.
In any case, I’ll be keeping an eye on Nooma. It seems like they’ve developed an attractive technique in search of a compelling application.