Read/Write DNA Devices – pt2
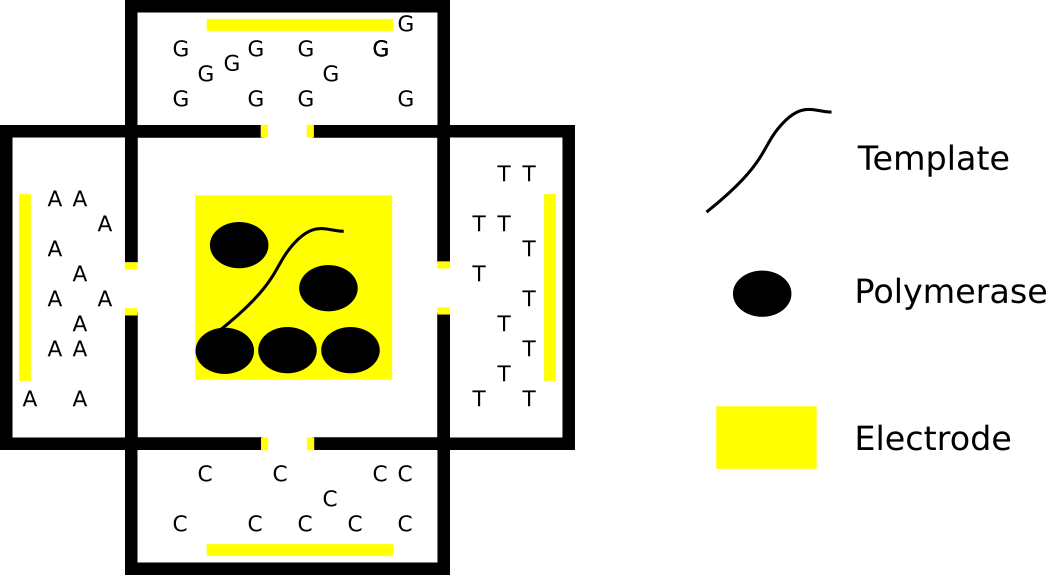
I previously wrote about a nanopore approach to creating a read/write DNA platform. In this post I decided to play with a bulk approach. The idea is to create a sequencing-by-synthesis system that does not require fluidics. All reagents can be preloaded, and is very cheap (the complete system would be in the 10USD range), but also very low throughput (maybe 100 sensors, though scaling maybe possible).
The idea is to move nucleotides around using electrophoresis [3], rather than fluid flow. In the diagram above the nucleotides sit in separate chambers [6], surrounding a central chamber where the template you are sequencing sits [5]. The template would need to be appropriately primed, this central chamber also contains polymerases (preloaded or otherwise).
The apertures connecting the nucleotides to the main chamber may also be surrounded by electrodes. These would be negatively charged to stop the nucleotides from migrating into the main chamber.
In order to sequence the template you would perform stepwise incorporation of each base, and detect the incorporation. For example, you would discharge the “A” aperture electrode. Negatively charge the electrode in the “A” chamber, and positively charge the electrode in the main chamber with respect to this. “A” nucleotides would then flow into the main chamber and incorporate into the template (if complementary to the current template position). After incorporation, unincorporated bases would need to be removed from the main chamber.
They could be removed in a number of ways. One method would be to reverse the polarity of the main chamber, and nucleotide chamber electrodes. This would return the nucleotides to their original chamber. For this to work, a sieving buffer would need to be used (as used in capillary electrophoresis sequencing for example). This would allow the single nucleotides to migrate quickly, and the template to migrate relatively slowly. As such the single nucleotides would migrate back to their original chamber [2] before the template (and polymerase) has had a chance to migrate out of the main chamber. After the nucleotides have returned, a charge could be applied to the main chamber to compensate for any movement of the template/polymerase and return them to the centre of the chamber.
Alternatively a nucleotide degrading enzyme as suggested by Ronaghi et al. in 1998 [1] could be used. In this scenario, new nucleotides would be moved in under electrophoretic flow but any unincorporated bases would be digested. You might need to use a higher concentration of nucleotides perhaps to ensure that some are incorporated before digestion.
Once incorporation has taken place, and free nucleotides have been removed [4] the incorporation can be detected. This maybe through the use of labelled nucleotides (fluorescence or otherwise). However it might also be possible to detect unlabelled molecules via the main chamber electrode, I would imagine the incorporation of additional nucleotides would result in a change in capacitance which could be detected. Or perhaps could be detected via multiple main chamber electrodes through other means.
So that’s how sequencing would work. Multiple incorporations would need to be detected through increased signal intensity (like other unterminated approaches) or through the use of reversible terminators.
It could be that users would want to use pre-amplified DNA with appropriate terminators. Or pre-diluted DNA such that only a single template is present (with appropriate primers). If single template is present, we’d also need to amplify it on device. A heater (TEC or otherwise) could be present on device to allow thermo-cycling to take place. The presented nucleotide chambers could be used, or an additional chamber containing a mixture of nucleotides.
So, there we go, that’s the sequencing side of things. In terms of construction a number of approaches could be used. I kind of envisage a platform based around commodity printed circuit boards (with ENIG plating perhaps). This would provide the electrodes and chambers would need to be built on top of this. Using a PCB it would probably be possible to scale out to ~100 chambers. Nucleotides and reagents could come preloaded. The sequencing “cell” would probably cost ~1 to 5USD. Associated electronics would likely not be very expensive, I could imagine this also being disposable. But if for example, picoamp current sensing and or CCD/photodiode acquisition is required over 100 channels, electronics could cost perhaps 100USD.
Some electrodes might come pre-energised to prevent nucleotide migration, which would be accomplished using a small embedded battery.
While it might be possible to scale out further than 100 sensors, a 100 sensor system would be targeted as a general purpose lab tool. It’s the kind of small scale, super cheap sequencer that doesn’t currently exist and which users could use as a routine part of protocols without spending 100s of dollars.
Writing
That’s the read side, but I believe it might be possible to use the same device for synthesis. In this scenario rather than using a normal polymerase, a template independent polymerase would be used (e.g. TdT). As before you’d selectively expose the polymerase to different bases to perform synthesis.
Obviously, without terminators it will be difficult to control the number of nucleotides incorporated accurately. However it maybe possible to control this to some degree through reagent concentration (and the nucleotide degrading approach above). There may also be other ways of preventing multiple bases from being incorporated.
However, if we have coarse grain control over the number of nucleotides incorporated (at least 1, less than 10 for example). Such a system could already have utility for data storage applications. In such a scenario we with use limited, or no information from the homopolymer length. We could for example using A->T transitions to represent 0, and G->C transitions to represent 1. A number of encoding are possible…
Notes
[1] Nucleotide degrading enzyme approach:
https://www.researchgate.net/publication/13579022_A_Sequencing_Method_Based_on_Real-Time_Pyrophosphate
[2] You might also want to move them back to a “waste” chamber, if there’s some possibility of mixing. This might require a larger supply of nucleotides.
[3] Or I guess electro-osmosis.
[4] Or even before they’ve been removed depending on the detection process.
[5] You can imagine a number of methods for loading the templates into the system. As a lab tool I’m kind of imagining that users would just load templates into a aperture with is sealed when shipped.
[6] Nucleotides could be preloaded. In an array system, nucleotide chambers might be connected to allow them to be loaded more easily. Or they could be “printed” etc.